Center for Computational Immunology, Computational Biology and Bioinformatics Program, Duke University, Durham, NC 27705, USA.
Bioinformatics. 2010 Apr 1;26(7):867-72. doi: 10.1093/bioinformatics/btq056. Epub 2010 Feb 9.
The inference of pre-mutation immunoglobulin (Ig) rearrangements is essential in the study of the antibody repertoires produced in response to infection, in B-cell neoplasms and in autoimmune disease. Often, there are several rearrangements that are nearly equivalent as candidates for a given Ig gene, but have different consequences in an analysis. Our aim in this article is to develop a probabilistic model of the rearrangement process and a Bayesian method for estimating posterior probabilities for the comparison of multiple plausible rearrangements.
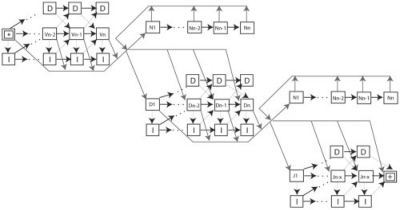
We have developed SoDA2, which is based on a Hidden Markov Model and used to compute the posterior probabilities of candidate rearrangements and to find those with the highest values among them. We validated the software on a set of simulated data, a set of clonally related sequences, and a group of randomly selected Ig heavy chains from Genbank. In most tests, SoDA2 performed better than other available software for the task. Furthermore, the output format has been redesigned, in part, to facilitate comparison of multiple solutions.
SoDA2 is available online at https://hippocrates.duhs.duke.edu/soda. Simulated sequences are available upon request.
在研究感染、B 细胞肿瘤和自身免疫性疾病中产生的抗体库时,推断免疫球蛋白(Ig)前突变重排是必不可少的。通常,有几个重排在候选 Ig 基因中几乎是等效的,但在分析中会有不同的结果。本文的目的是开发一种重排过程的概率模型和一种贝叶斯方法,用于比较多个合理重排的后验概率。
我们开发了 SoDA2,它基于隐马尔可夫模型,用于计算候选重排的后验概率,并从中找到最高值的那些。我们在一组模拟数据、一组克隆相关序列和一组从 Genbank 中随机选择的 Ig 重链上验证了该软件。在大多数测试中,SoDA2 在该任务上的表现优于其他可用软件。此外,输出格式已部分重新设计,以方便比较多个解决方案。
SoDA2 可在 https://hippocrates.duhs.duke.edu/soda 在线获取。模拟序列可根据要求提供。