CEINGE-Biotecnologie Avanzate, Napoli, Dipartimento di Scienze Biologiche ed Ambientali, Università del Sannio, Benevento, Naples, Italy.
J Inherit Metab Dis. 2010 Dec;33 Suppl 3(Suppl 3):S91-4. doi: 10.1007/s10545-009-9028-3. Epub 2010 Feb 16.
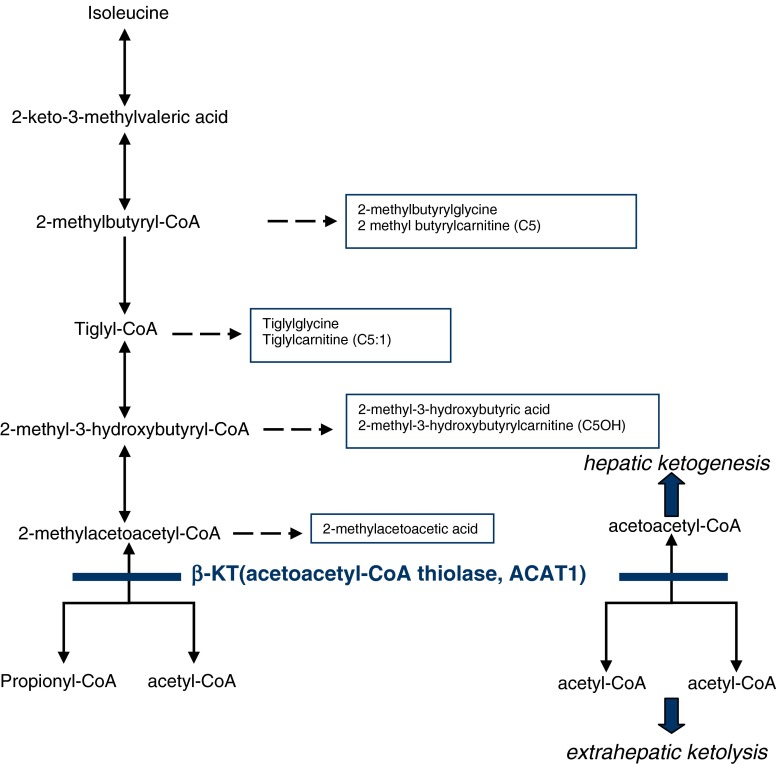
A pilot expanded newborn screening programme to detect inherited metabolic disorders by means of liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) began in the Campania region, southern Italy, in 2007. By October 2009, >8,800 dried blood samples on filter paper from 11 hospitals had been screened. Within this screening programme, we identified a case of mitochondrial acetoacetyl-coenzyme A (CoA) thiolase deficiency [β-ketothiolase (β-KT) deficiency] by analysing the acylcarnitine profile from a dried blood spot with LC-MS/MS. Gas chromatography coupled with mass spectrometry analysis of urinary organic acids and LC-MS/MS analysis of urinary acylcarnitines were in line with this disorder. In fact, concentrations were well beyond the cut-off values of tiglyl carnitine, 3-hydroxybutyrylcarnitine and 2-methyl-3-hydroxybutyrylcarnitine, 2-methyl-3-hydroxybutyric acid and tiglyl glycine. The absence of 2-methylacetoacetic acid in urine may be attributed to: (i) the instability of this β-ketoacid because it undergoes spontaneous decarboxylation to 2-butanone, which is highly volatile and thus difficult to detect, and (ii) the good health of the patient in the first days of life. β-KT deficiency was subsequently diagnosed in the patient's older sister, who showed increased levels of the same metabolites but also small amounts of 2-methylacetoacetic acid, which is considered a key marker for β-KT diagnosis. Genomic analysis revealed mutation c.1189C >G in exon 12 of the ACAT1 gene, which results in a severe defect because of the p.H397D amino acid change in both alleles of both patients.
意大利坎帕尼亚地区于 2007 年启动了一项通过液相色谱串联质谱法(LC-MS/MS)扩大新生儿筛查计划,以检测遗传性代谢疾病。截至 2009 年 10 月,已有来自 11 家医院的超过 8800 份滤纸干血斑样本接受了筛查。在这个筛查项目中,我们通过 LC-MS/MS 分析滤纸干血斑中的酰基肉碱谱,发现了一例线粒体乙酰乙酰辅酶 A(CoA)硫解酶缺乏症(β-酮硫解酶[β-KT]缺乏症)。气相色谱-质谱联用分析尿液有机酸和 LC-MS/MS 分析尿液酰基肉碱与这种疾病一致。事实上,浓度远远超过了琥珀酰肉碱、3-羟基丁酰肉碱和 2-甲基-3-羟基丁酰肉碱、2-甲基-3-羟基丁酸和丁酰甘氨酸的截断值。尿液中 2-甲基乙酰乙酸的缺失可能归因于:(i)β-酮酸的不稳定性,因为它会自发脱羧生成 2-丁酮,2-丁酮具有很高的挥发性,因此难以检测;(ii)患者在生命的最初几天身体状况良好。随后在患者的姐姐中诊断出β-KT 缺乏症,她的同种代谢物水平升高,但也有少量 2-甲基乙酰乙酸,这被认为是β-KT 诊断的关键标志物。基因组分析显示 ACAT1 基因第 12 外显子的 c.1189C > G 突变,导致两个患者的两个等位基因都发生了 p.H397D 氨基酸变化,从而导致严重缺陷。