Dipartimento di Medicina Molecolare e Biotecnologie Mediche, Università degli Studi di Napoli "Federico II", 80131 Naples, Italy.
CEINGE-Biotecnologie Avanzate s.c.ar.l., 80145 Naples, Italy.
Int J Mol Sci. 2018 Nov 13;19(11):3580. doi: 10.3390/ijms19113580.
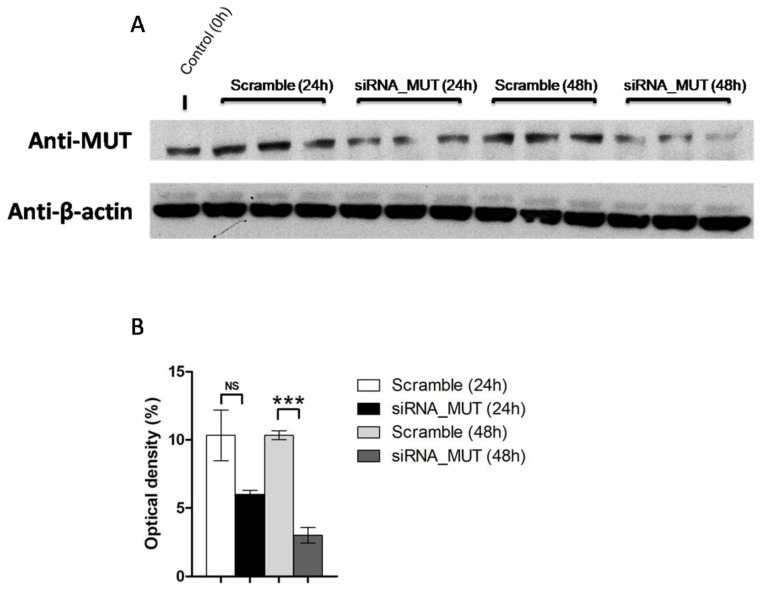
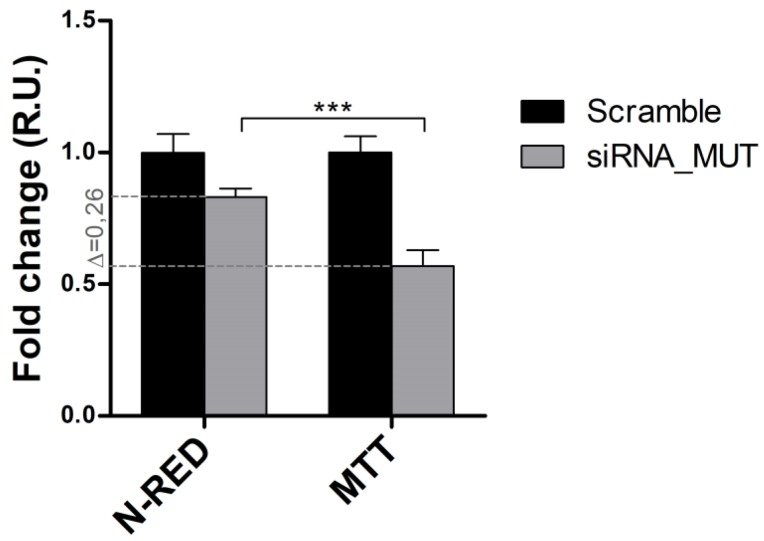
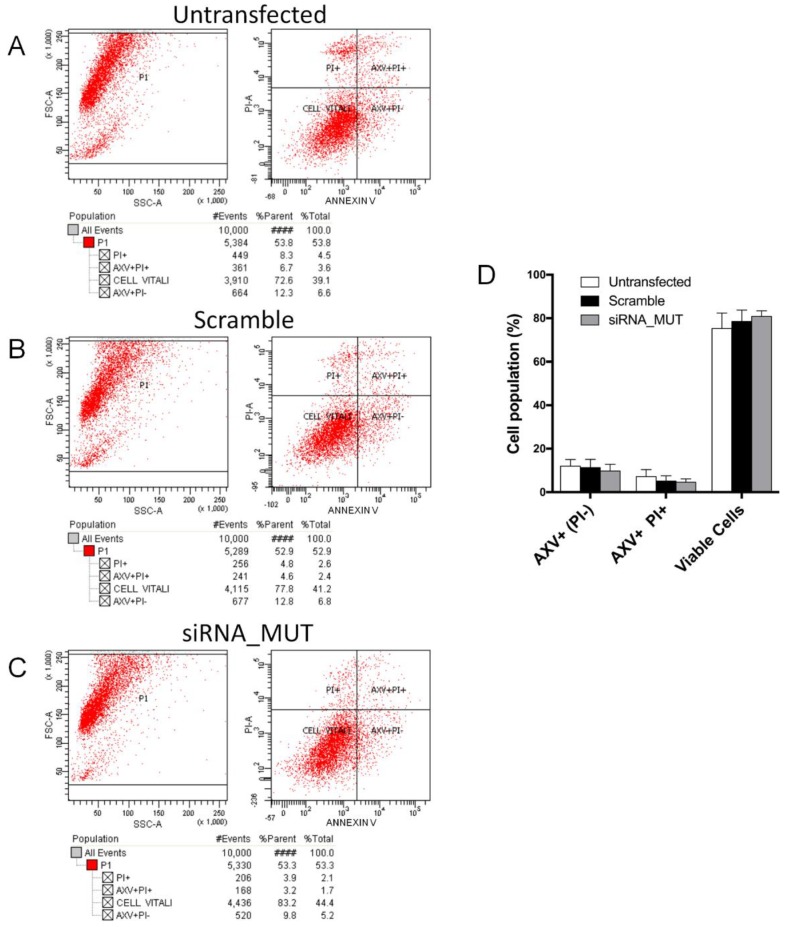
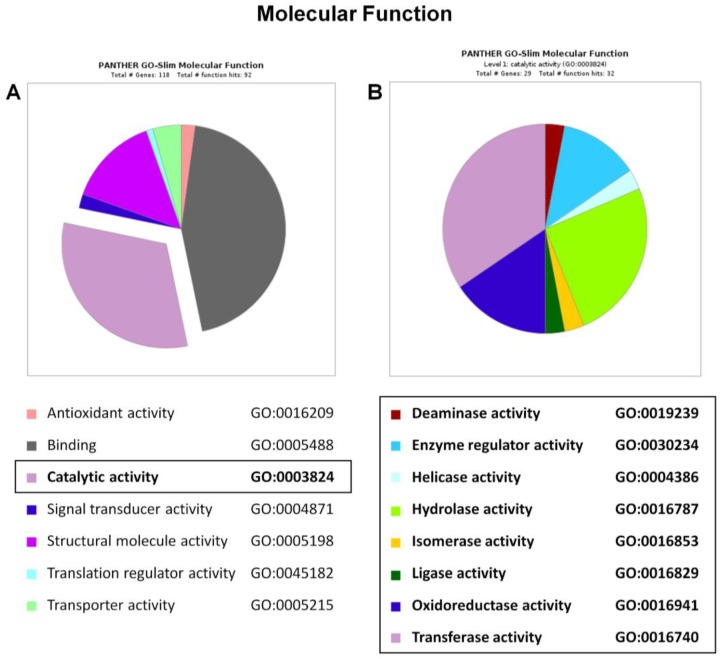
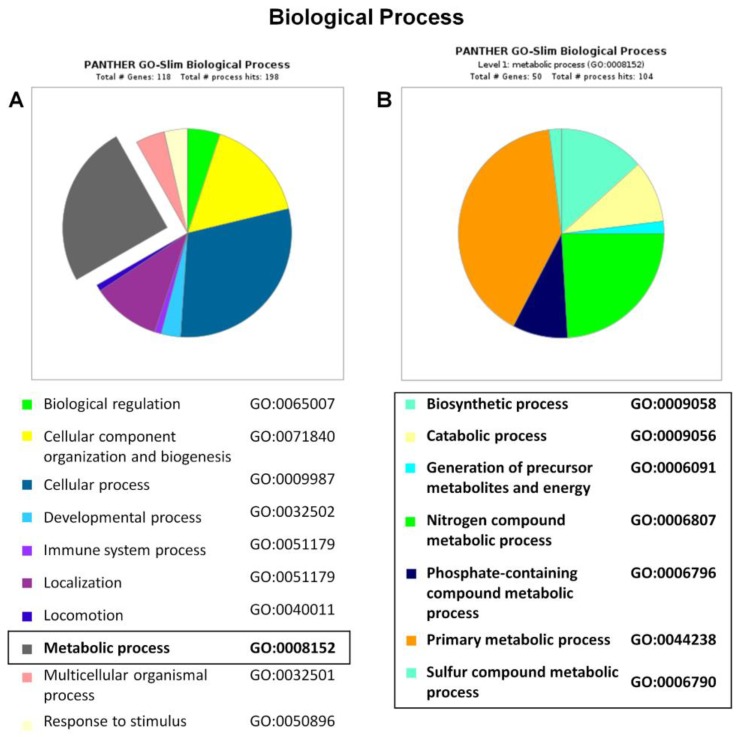
Methylmalonic acidemias (MMAs) are inborn errors of metabolism due to the deficient activity of methylmalonyl-CoA mutase (MUT). MUT catalyzes the formation of succinyl-CoA from methylmalonyl-CoA, produced from propionyl-CoA catabolism and derived from odd chain fatty acids β-oxidation, cholesterol, and branched-chain amino acids degradation. Increased methylmalonyl-CoA levels allow for the presymptomatic diagnosis of the disease, even though no approved therapies exist. MMA patients show hyperammonemia, ketoacidosis, lethargy, respiratory distress, cognitive impairment, and hepatomegaly. The long-term consequences concern neurologic damage and terminal kidney failure, with little chance of survival. The cellular pathways affected by MUT deficiency were investigated using a quantitative proteomics approach on a cellular model of MUT knockdown. Currently, a consistent reduction of the MUT protein expression was obtained in the neuroblastoma cell line (SH-SY5Y) by using small-interfering RNA (siRNA) directed against an MUT transcript (MUT siRNA). The MUT absence did not affect the cell viability and apoptotic process in SH-SY5Y. In the present study, we evaluate and quantify the alterations in the protein expression profile as a consequence of MUT-silencing by a mass spectrometry-based label-free quantitative analysis, using two different quantitative strategies. Both quantitative methods allowed us to observe that the expression of the proteins involved in mitochondrial oxido-reductive homeostasis balance was affected by MUT deficiency. The alterated functional mitochondrial activity was observed in siRNA_MUT cells cultured with a propionate-supplemented medium. Finally, alterations in the levels of proteins involved in the metabolic pathways, like carbohydrate metabolism and lipid metabolism, were found.
甲基丙二酸血症(MMAs)是由于甲基丙二酰辅酶 A 变位酶(MUT)活性缺乏导致的先天性代谢缺陷。MUT 催化甲基丙二酰辅酶 A 转化为琥珀酰辅酶 A,该反应由丙酰辅酶 A 分解代谢产生,并来自奇数链脂肪酸β氧化、胆固醇和支链氨基酸降解。甲基丙二酰辅酶 A 水平升高可用于疾病的早期诊断,尽管目前尚无批准的治疗方法。MMA 患者表现为高血氨、酮酸中毒、嗜睡、呼吸窘迫、认知障碍和肝肿大。长期后果涉及神经系统损伤和终末期肾衰竭,几乎没有生存机会。使用定量蛋白质组学方法在 MUT 敲低的细胞模型上研究了受 MUT 缺乏影响的细胞途径。目前,通过使用针对 MUT 转录本(MUT siRNA)的小干扰 RNA(siRNA),在神经母细胞瘤细胞系(SH-SY5Y)中一致降低了 MUT 蛋白表达。MUT 缺失不影响 SH-SY5Y 中的细胞活力和凋亡过程。在本研究中,我们通过基于质谱的无标记定量分析,使用两种不同的定量策略,评估和量化由于 MUT 沉默导致的蛋白质表达谱的变化。这两种定量方法都使我们能够观察到参与线粒体氧化还原稳态平衡的蛋白质的表达因 MUT 缺乏而受到影响。在含有丙酸盐补充培养基中培养的 siRNA_MUT 细胞中观察到功能失调的线粒体活性改变。最后,还发现了参与代谢途径的蛋白质水平的改变,如碳水化合物代谢和脂质代谢。