Life Technologies Inc., Foster City, California, United States of America.
PLoS One. 2010 Feb 19;5(2):e9317. doi: 10.1371/journal.pone.0009317.
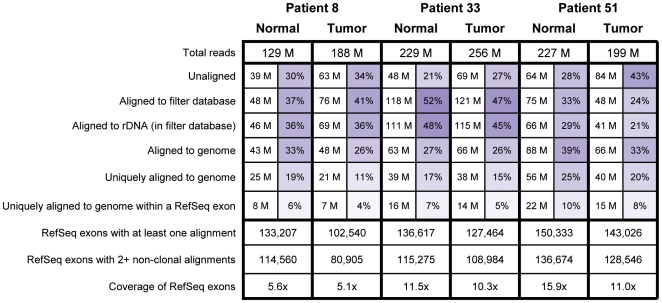
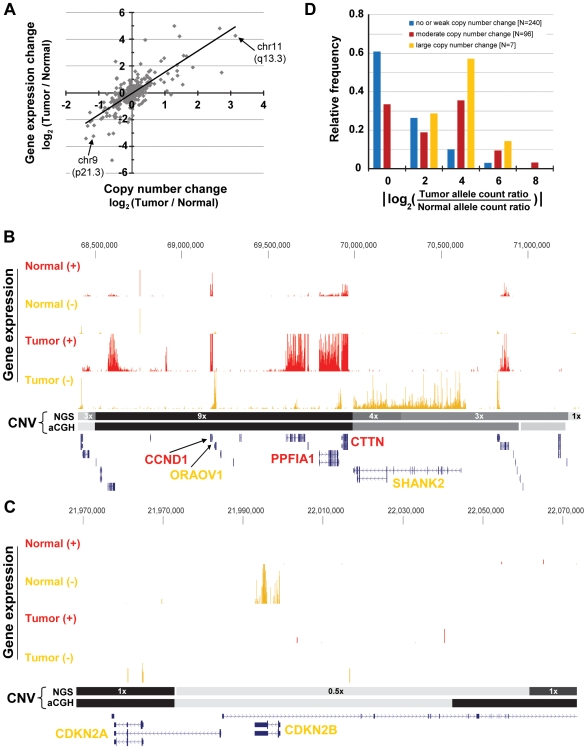
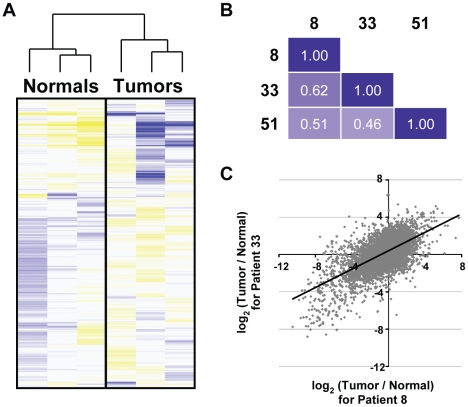
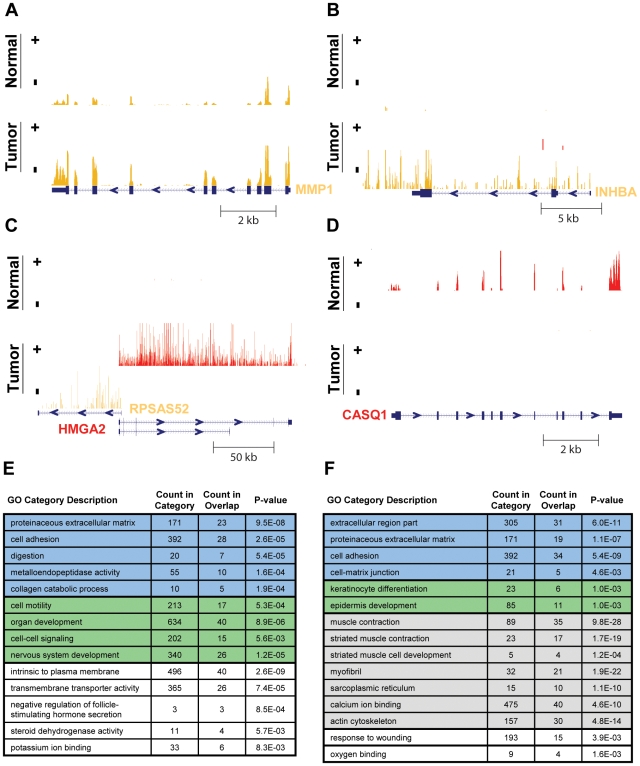
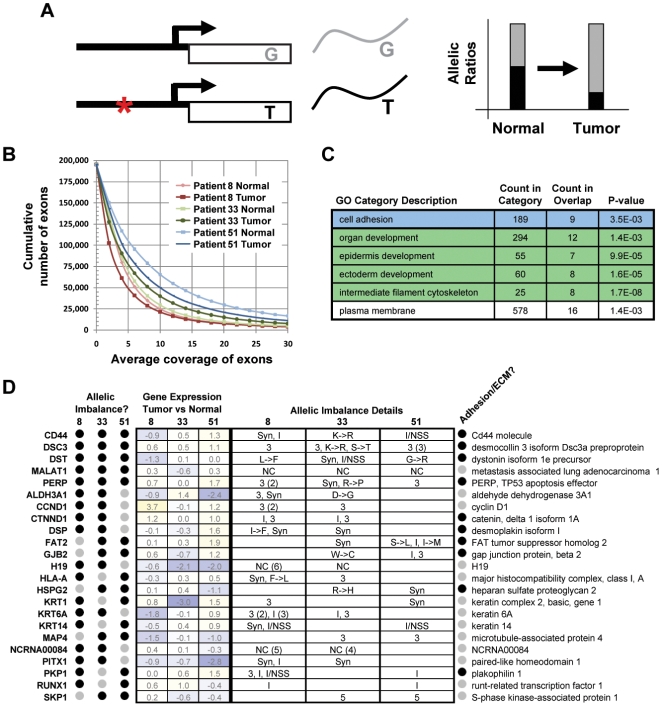
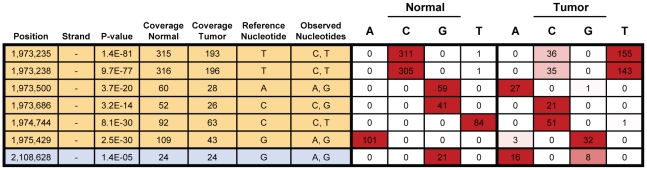
Due to growing throughput and shrinking cost, massively parallel sequencing is rapidly becoming an attractive alternative to microarrays for the genome-wide study of gene expression and copy number alterations in primary tumors. The sequencing of transcripts (RNA-Seq) should offer several advantages over microarray-based methods, including the ability to detect somatic mutations and accurately measure allele-specific expression. To investigate these advantages we have applied a novel, strand-specific RNA-Seq method to tumors and matched normal tissue from three patients with oral squamous cell carcinomas. Additionally, to better understand the genomic determinants of the gene expression changes observed, we have sequenced the tumor and normal genomes of one of these patients. We demonstrate here that our RNA-Seq method accurately measures allelic imbalance and that measurement on the genome-wide scale yields novel insights into cancer etiology. As expected, the set of genes differentially expressed in the tumors is enriched for cell adhesion and differentiation functions, but, unexpectedly, the set of allelically imbalanced genes is also enriched for these same cancer-related functions. By comparing the transcriptomic perturbations observed in one patient to his underlying normal and tumor genomes, we find that allelic imbalance in the tumor is associated with copy number mutations and that copy number mutations are, in turn, strongly associated with changes in transcript abundance. These results support a model in which allele-specific deletions and duplications drive allele-specific changes in gene expression in the developing tumor.
由于通量的增加和成本的降低,大规模平行测序技术迅速成为一种有吸引力的选择,可替代微阵列进行原发性肿瘤的基因表达和拷贝数改变的全基因组研究。转录本测序(RNA-Seq)应该比基于微阵列的方法具有几个优势,包括检测体细胞突变和准确测量等位基因特异性表达的能力。为了研究这些优势,我们应用了一种新的、链特异性的 RNA-Seq 方法,对三例口腔鳞状细胞癌患者的肿瘤和匹配的正常组织进行了研究。此外,为了更好地理解观察到的基因表达变化的基因组决定因素,我们对其中一名患者的肿瘤和正常基因组进行了测序。我们在这里证明,我们的 RNA-Seq 方法能够准确地测量等位基因失衡,并且在全基因组范围内进行测量可以为癌症病因学提供新的见解。正如预期的那样,在肿瘤中差异表达的基因集富集了细胞黏附和分化功能,但出乎意料的是,等位基因失衡的基因集也富集了这些与癌症相关的功能。通过将一名患者的转录组扰动与其潜在的正常和肿瘤基因组进行比较,我们发现肿瘤中的等位基因失衡与拷贝数突变有关,而拷贝数突变又与转录物丰度的变化密切相关。这些结果支持了这样一种模型,即等位基因特异性缺失和重复驱动了肿瘤发育过程中基因表达的等位基因特异性变化。