Okada Yoshifumi, Inoue Terufumi
College of Information and Systems, Muroran Institute of Technology, 27-1, Mizumoto-cho, Muroran 050-8585, Japan.
Bioinformation. 2009 Oct 11;4(4):134-7. doi: 10.6026/97320630004134.
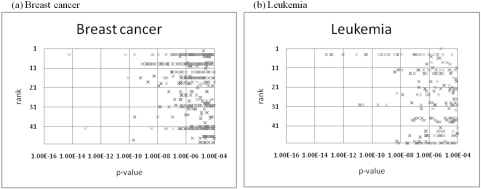
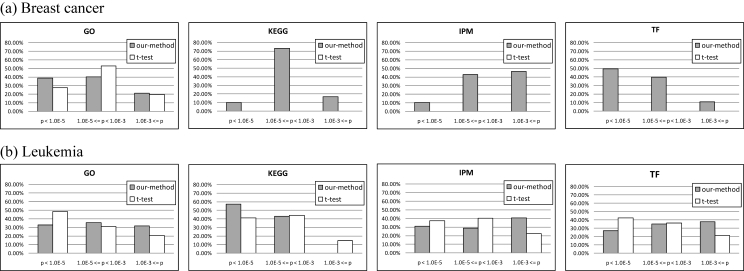
Identifying biologically useful genes from massive gene expression data is a critical issue in DNA microarray data analysis. Recent studies on gene module discovery have shown a substantial effect on identifying transcriptional regulatory networks involved in complex diseases for different sample subsets. These have targeted a single disease class, but discovering discriminative modules in different classes has remained to be addressed. In this paper, we propose a novel method that can discover differentially expressed gene modules from two-class DNA microarray data. The proposed method is applied to breast cancer and leukemia datasets, and the biological functions of the extracted modules are evaluated by functional enrichment analysis. As a result, we show that our method can extract genes well reflecting known biological functions compared to a traditional t-test-based approach.
从海量基因表达数据中识别具有生物学意义的基因是DNA微阵列数据分析中的关键问题。近期关于基因模块发现的研究表明,对于不同样本子集,在识别复杂疾病相关的转录调控网络方面有显著成效。这些研究针对的是单一疾病类别,但在不同类别中发现有区分性的模块这一问题仍有待解决。在本文中,我们提出了一种新方法,该方法能够从两类DNA微阵列数据中发现差异表达的基因模块。所提出的方法应用于乳腺癌和白血病数据集,并通过功能富集分析评估所提取模块的生物学功能。结果表明,与传统的基于t检验的方法相比,我们的方法能够更好地提取出反映已知生物学功能的基因。