International Crops Research Institute for the Semi-Arid Tropics (ICRISAT), Patancheru, Greater Hyderabad, Andhra Pradesh, India.
BMC Plant Biol. 2010 Mar 11;10:45. doi: 10.1186/1471-2229-10-45.
Pigeonpea (Cajanus cajan (L.) Millsp) is one of the major grain legume crops of the tropics and subtropics, but biotic stresses [Fusarium wilt (FW), sterility mosaic disease (SMD), etc.] are serious challenges for sustainable crop production. Modern genomic tools such as molecular markers and candidate genes associated with resistance to these stresses offer the possibility of facilitating pigeonpea breeding for improving biotic stress resistance. Availability of limited genomic resources, however, is a serious bottleneck to undertake molecular breeding in pigeonpea to develop superior genotypes with enhanced resistance to above mentioned biotic stresses. With an objective of enhancing genomic resources in pigeonpea, this study reports generation and analysis of comprehensive resource of FW- and SMD- responsive expressed sequence tags (ESTs).
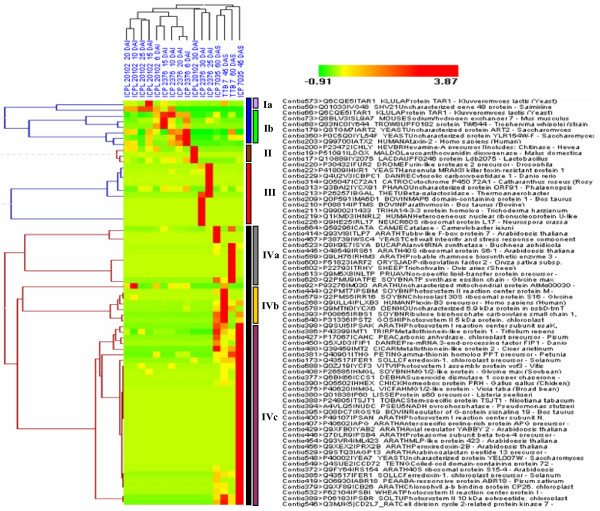
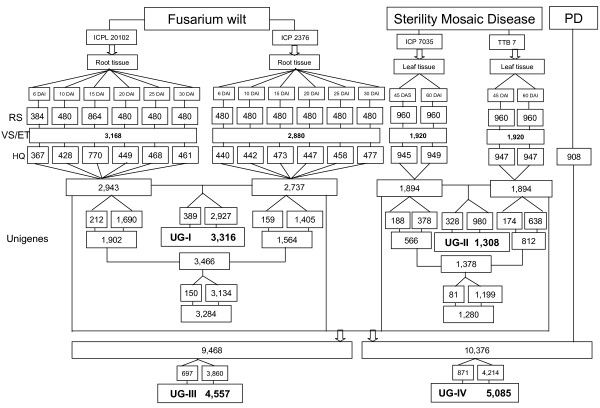
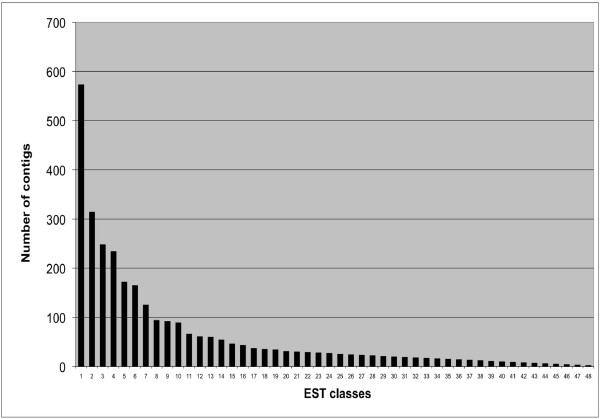
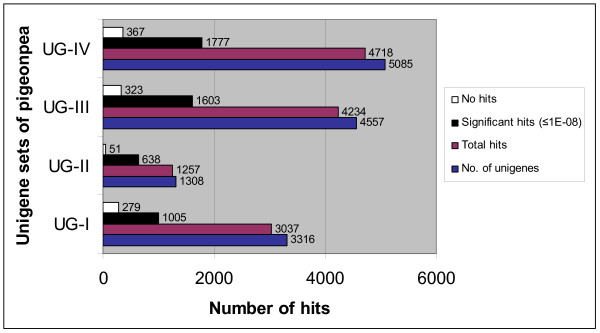
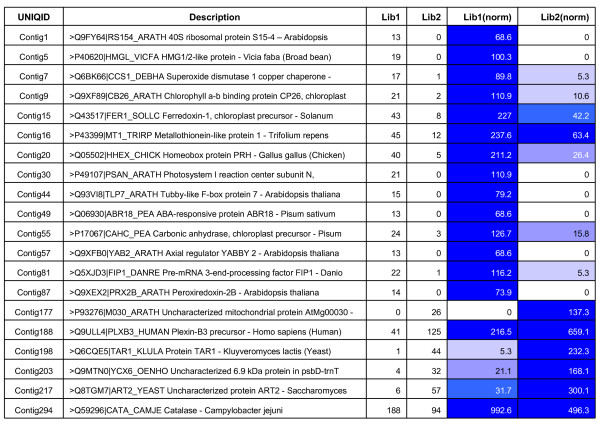
A total of 16 cDNA libraries were constructed from four pigeonpea genotypes that are resistant and susceptible to FW ('ICPL 20102' and 'ICP 2376') and SMD ('ICP 7035' and 'TTB 7') and a total of 9,888 (9,468 high quality) ESTs were generated and deposited in dbEST of GenBank under accession numbers GR463974 to GR473857 and GR958228 to GR958231. Clustering and assembly analyses of these ESTs resulted into 4,557 unique sequences (unigenes) including 697 contigs and 3,860 singletons. BLASTN analysis of 4,557 unigenes showed a significant identity with ESTs of different legumes (23.2-60.3%), rice (28.3%), Arabidopsis (33.7%) and poplar (35.4%). As expected, pigeonpea ESTs are more closely related to soybean (60.3%) and cowpea ESTs (43.6%) than other plant ESTs. Similarly, BLASTX similarity results showed that only 1,603 (35.1%) out of 4,557 total unigenes correspond to known proteins in the UniProt database (<or= 1E-08). Functional categorization of the annotated unigenes sequences showed that 153 (3.3%) genes were assigned to cellular component category, 132 (2.8%) to biological process, and 132 (2.8%) in molecular function. Further, 19 genes were identified differentially expressed between FW- responsive genotypes and 20 between SMD- responsive genotypes. Generated ESTs were compiled together with 908 ESTs available in public domain, at the time of analysis, and a set of 5,085 unigenes were defined that were used for identification of molecular markers in pigeonpea. For instance, 3,583 simple sequence repeat (SSR) motifs were identified in 1,365 unigenes and 383 primer pairs were designed. Assessment of a set of 84 primer pairs on 40 elite pigeonpea lines showed polymorphism with 15 (28.8%) markers with an average of four alleles per marker and an average polymorphic information content (PIC) value of 0.40. Similarly, in silico mining of 133 contigs with >or= 5 sequences detected 102 single nucleotide polymorphisms (SNPs) in 37 contigs. As an example, a set of 10 contigs were used for confirming in silico predicted SNPs in a set of four genotypes using wet lab experiments. Occurrence of SNPs were confirmed for all the 6 contigs for which scorable and sequenceable amplicons were generated. PCR amplicons were not obtained in case of 4 contigs. Recognition sites for restriction enzymes were identified for 102 SNPs in 37 contigs that indicates possibility of assaying SNPs in 37 genes using cleaved amplified polymorphic sequences (CAPS) assay.
The pigeonpea EST dataset generated here provides a transcriptomic resource for gene discovery and development of functional markers associated with biotic stress resistance. Sequence analyses of this dataset have showed conservation of a considerable number of pigeonpea transcripts across legume and model plant species analysed as well as some putative pigeonpea specific genes. Validation of identified biotic stress responsive genes should provide candidate genes for allele mining as well as candidate markers for molecular breeding.

木豆(Cajanus cajan(L.)Millsp)是热带和亚热带地区主要的粮食豆类作物之一,但生物胁迫[枯萎病(FW)、不育嵌合体病(SMD)等]是可持续作物生产的严重挑战。现代基因组工具,如与这些胁迫抗性相关的分子标记和候选基因,为通过改善生物胁迫抗性进行木豆育种提供了可能。然而,有限的基因组资源的可用性是在木豆中进行分子育种以开发具有增强的生物胁迫抗性的优良基因型的严重瓶颈。本研究的目的是增强木豆的基因组资源,报告 FW 和 SMD 反应表达序列标签(EST)综合资源的生成和分析。
从对 FW('ICPL 20102'和'ICP 2376')和 SMD('ICP 7035'和'TTB 7')具有抗性和敏感性的四个木豆基因型中构建了总共 16 个 cDNA 文库,共产生并保存在 GenBank 的 dbEST 中,登录号为 GR463974 至 GR473857 和 GR958228 至 GR958231。这些 EST 的聚类和组装分析产生了 4557 个独特的序列(unigenes),包括 697 个 contigs 和 3860 个 singletons。对 4557 个 unigenes的 BLASTN 分析显示,与不同豆科植物(23.2-60.3%)、水稻(28.3%)、拟南芥(33.7%)和杨树(35.4%)的 EST 有显著的同一性。正如预期的那样,与其他植物 EST 相比,木豆 EST 与大豆(60.3%)和豇豆 EST(43.6%)的亲缘关系更密切。同样,BLASTX 相似性结果表明,4557 个总 unigenes中只有 1603 个(35.1%)与 UniProt 数据库中的已知蛋白相对应(<或= 1E-08)。注释 unigenes 序列的功能分类表明,153 个(3.3%)基因被分配到细胞成分类别,132 个(2.8%)到生物过程,132 个(2.8%)到分子功能。此外,在 FW 反应基因型之间鉴定到 19 个差异表达的基因,在 SMD 反应基因型之间鉴定到 20 个基因。生成的 EST 与当时可公开获得的 908 个 EST 一起进行编译,定义了一组 5085 个 unigenes,用于鉴定木豆中的分子标记。例如,在 1365 个 unigenes中鉴定出 3583 个简单序列重复(SSR)基序,并设计了 383 对引物。对 40 个木豆优良品系的 84 对引物的评估显示,15 对(28.8%)标记具有平均每个标记 4 个等位基因和平均多态信息含量(PIC)值为 0.40。同样,通过 >或= 5 个序列对 133 个 contigs 的计算机挖掘检测到 37 个 contigs 中的 102 个单核苷酸多态性(SNP)。例如,使用四个基因型的湿实验室实验,对一组 10 个 contigs 进行了确认计算机预测的 SNP。所有 6 个可评分和可测序的扩增子的 contigs 都证实了 SNP 的发生。对于没有产生 PCR 扩增子的 4 个 contigs,识别到限制酶的识别位点。在 37 个 contigs 中,102 个 SNP 表明可以使用切割扩增多态性序列(CAPS)检测 37 个基因中的 SNP。
这里生成的木豆 EST 数据集为基因发现和与生物胁迫抗性相关的功能标记的开发提供了转录组资源。对该数据集的序列分析表明,在分析的豆科植物和模式植物物种中,相当数量的木豆转录本具有保守性,以及一些假定的木豆特异性基因。鉴定出的生物胁迫反应基因的验证应该为等位基因挖掘提供候选基因,并为分子育种提供候选标记。