National Research Centre on Plant Biotechnology, Indian Agricultural Research Institute, New Delhi 110 012, India.
BMC Plant Biol. 2011 Jan 20;11:17. doi: 10.1186/1471-2229-11-17.
Pigeonpea [Cajanus cajan (L.) Millspaugh], one of the most important food legumes of semi-arid tropical and subtropical regions, has limited genomic resources, particularly expressed sequence based (genic) markers. We report a comprehensive set of validated genic simple sequence repeat (SSR) markers using deep transcriptome sequencing, and its application in genetic diversity analysis and mapping.
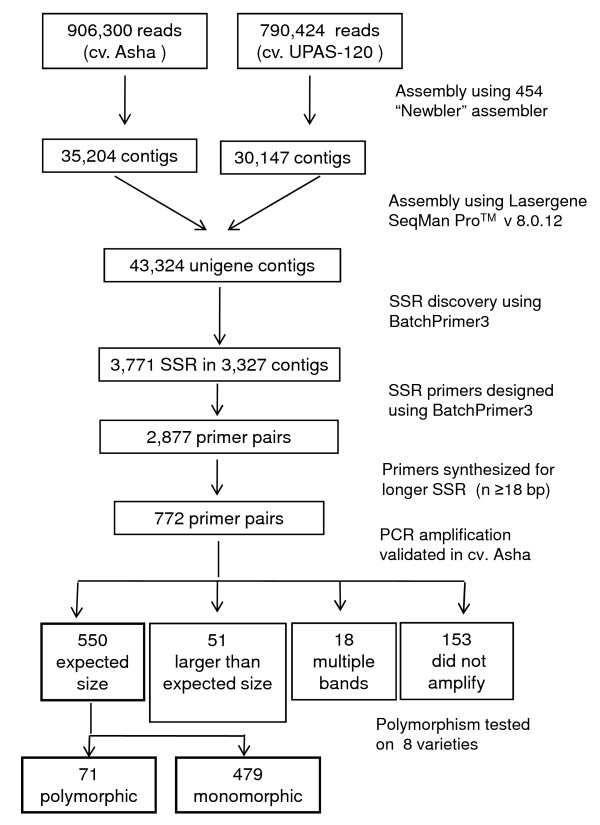
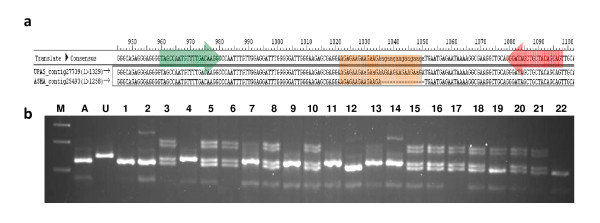
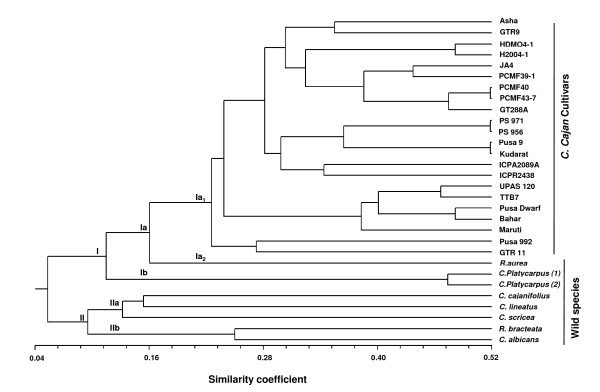
In this study, 43,324 transcriptome shotgun assembly unigene contigs were assembled from 1.696 million 454 GS-FLX sequence reads of separate pooled cDNA libraries prepared from leaf, root, stem and immature seed of two pigeonpea varieties, Asha and UPAS 120. A total of 3,771 genic-SSR loci, excluding homopolymeric and compound repeats, were identified; of which 2,877 PCR primer pairs were designed for marker development. Dinucleotide was the most common repeat motif with a frequency of 60.41%, followed by tri- (34.52%), hexa- (2.62%), tetra- (1.67%) and pentanucleotide (0.76%) repeat motifs. Primers were synthesized and tested for 772 of these loci with repeat lengths of ≥ 18 bp. Of these, 550 markers were validated for consistent amplification in eight diverse pigeonpea varieties; 71 were found to be polymorphic on agarose gel electrophoresis. Genetic diversity analysis was done on 22 pigeonpea varieties and eight wild species using 20 highly polymorphic genic-SSR markers. The number of alleles at these loci ranged from 4-10 and the polymorphism information content values ranged from 0.46 to 0.72. Neighbor-joining dendrogram showed distinct separation of the different groups of pigeonpea cultivars and wild species. Deep transcriptome sequencing of the two parental lines helped in silico identification of polymorphic genic-SSR loci to facilitate the rapid development of an intra-species reference genetic map, a subset of which was validated for expected allelic segregation in the reference mapping population.
We developed 550 validated genic-SSR markers in pigeonpea using deep transcriptome sequencing. From these, 20 highly polymorphic markers were used to evaluate the genetic relationship among species of the genus Cajanus. A comprehensive set of genic-SSR markers was developed as an important genomic resource for diversity analysis and genetic mapping in pigeonpea.
木豆[Cajanus cajan (L.) Millspaugh]是半干旱热带和亚热带地区最重要的食用豆类之一,其基因组资源有限,特别是基于表达序列的(基因)标记。我们报告了一组使用深度转录组测序验证的综合基因简单重复序列(SSR)标记,并将其应用于遗传多样性分析和作图。
在这项研究中,从两个木豆品种 Asha 和 UPAS 120 的叶、根、茎和未成熟种子的单独混合 cDNA 文库中,从 1696000 个 454GS-FLX 序列读取中组装了 43324 个转录组 shotgun 组装的 unigene 片段。共鉴定出 3771 个基因 SSR 位点,不包括同聚和复合重复,其中 2877 对 PCR 引物被设计用于标记开发。二核苷酸是最常见的重复基序,频率为 60.41%,其次是三核苷酸(34.52%)、六核苷酸(2.62%)、四核苷酸(1.67%)和五核苷酸(0.76%)重复基序。合成并测试了这些长度大于 18 个 bp 的重复序列中的 772 个重复序列的引物。其中,550 个标记在 8 个不同的木豆品种中得到了一致的扩增验证;71 个标记在琼脂糖凝胶电泳中显示出多态性。使用 20 个高度多态性的基因 SSR 标记对 22 个木豆品种和 8 个野生种进行了遗传多样性分析。这些位点的等位基因数为 4-10,多态信息含量值为 0.46-0.72。邻接法聚类树状图显示了不同木豆品种和野生种的明显分离。对两个亲本系进行深度转录组测序有助于在计算机上鉴定多态性基因 SSR 位点,以促进种内参考遗传图谱的快速开发,其中一部分在参考作图群体中验证了预期的等位基因分离。
我们使用深度转录组测序在木豆中开发了 550 个经过验证的基因 SSR 标记。其中 20 个高度多态性标记用于评估该属物种之间的遗传关系。综合的基因 SSR 标记集作为多样性分析和木豆遗传作图的重要基因组资源得到了开发。