Department of Genome Sciences University of Washington, Seattle, WA, USA.
BMC Bioinformatics. 2010 Mar 18;11:144. doi: 10.1186/1471-2105-11-144.
The prediction of protein-protein interactions is an important step toward the elucidation of protein functions and the understanding of the molecular mechanisms inside the cell. While experimental methods for identifying these interactions remain costly and often noisy, the increasing quantity of solved 3D protein structures suggests that in silico methods to predict interactions between two protein structures will play an increasingly important role in screening candidate interacting pairs. Approaches using the knowledge of the structure are presumably more accurate than those based on sequence only. Approaches based on docking protein structures solve a variant of this problem, but these methods remain very computationally intensive and will not scale in the near future to the detection of interactions at the level of an interactome, involving millions of candidate pairs of proteins.
Here, we describe a computational method to predict efficiently in silico whether two protein structures interact. This yes/no question is presumably easier to answer than the standard protein docking question, "How do these two protein structures interact?" Our approach is to discriminate between interacting and non-interacting protein pairs using a statistical pattern recognition method known as a support vector machine (SVM). We demonstrate that our structure-based method performs well on this task and scales well to the size of an interactome.
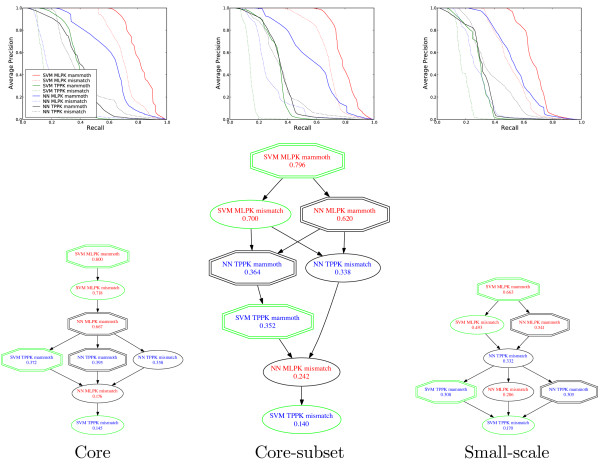
The use of structure information for the prediction of protein interaction yields significantly better performance than other sequence-based methods. Among structure-based classifiers, the SVM algorithm, combined with the metric learning pairwise kernel and the MAMMOTH kernel, performs best in our experiments.
预测蛋白质-蛋白质相互作用是阐明蛋白质功能和理解细胞内分子机制的重要步骤。虽然鉴定这些相互作用的实验方法仍然昂贵且常常存在噪声,但解决的 3D 蛋白质结构的数量不断增加表明,预测两个蛋白质结构之间相互作用的计算方法将在筛选候选相互作用对中发挥越来越重要的作用。基于结构知识的方法可能比仅基于序列的方法更准确。基于对接蛋白质结构的方法解决了这个问题的一个变体,但这些方法仍然非常计算密集,并且在不久的将来不会扩展到检测相互作用的水平,涉及数百万对候选蛋白质。
在这里,我们描述了一种有效地预测蛋白质结构是否相互作用的计算方法。这个是或否的问题可能比标准的蛋白质对接问题“这两个蛋白质结构如何相互作用?”更容易回答。我们的方法是使用称为支持向量机 (SVM) 的统计模式识别方法来区分相互作用和非相互作用的蛋白质对。我们证明我们基于结构的方法在这项任务上表现良好,并且可以很好地扩展到相互作用组的大小。
使用结构信息预测蛋白质相互作用的性能明显优于其他基于序列的方法。在基于结构的分类器中,SVM 算法与度量学习成对核和 MAMMOTH 核相结合,在我们的实验中表现最佳。