Departments of Cell Biology and Neurology, Center for Neurodegenerative Diseases, Emory University School of Medicine, Atlanta, GA 30322, USA.
Mol Brain. 2010 Mar 26;3:10. doi: 10.1186/1756-6606-3-10.
Synaptic defects represent a major mechanism underlying altered brain functions of patients suffering Alzheimer's disease (AD) 123. An increasing body of work indicates that the oligomeric forms of beta-amyloid (Abeta) molecules exert profound inhibition on synaptic functions and can cause a significant loss of neurotransmitter receptors from the postsynaptic surface, but the underlying mechanisms remain poorly understood. In this study, we investigated a potential contribution of mitochondria to Abeta inhibition of AMPA receptor (AMPAR) trafficking.
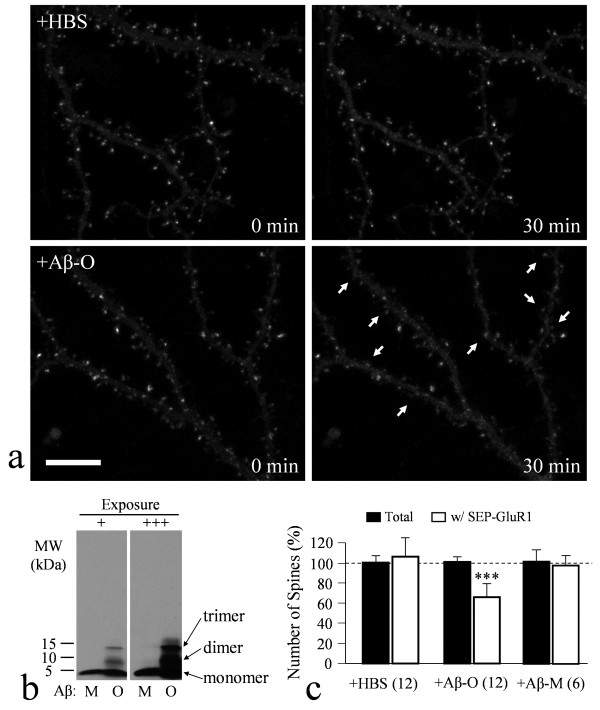
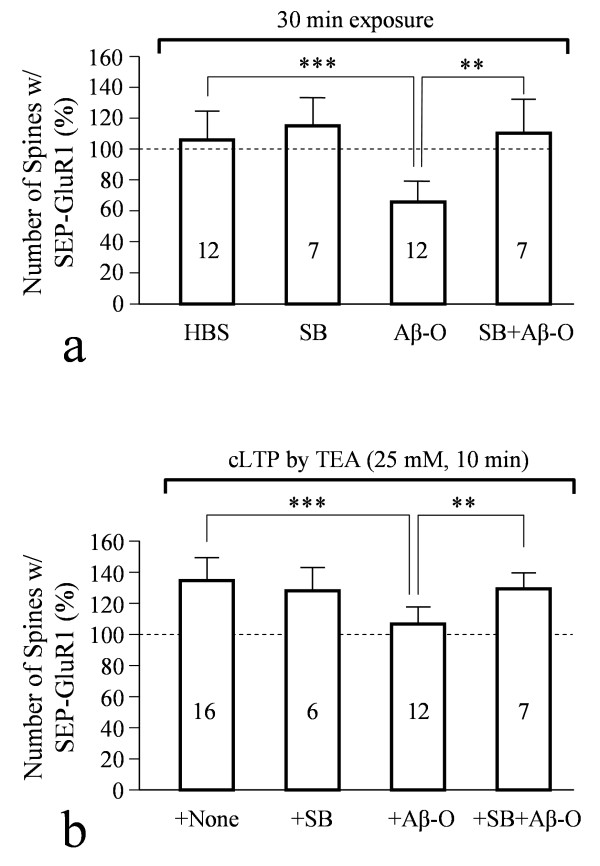
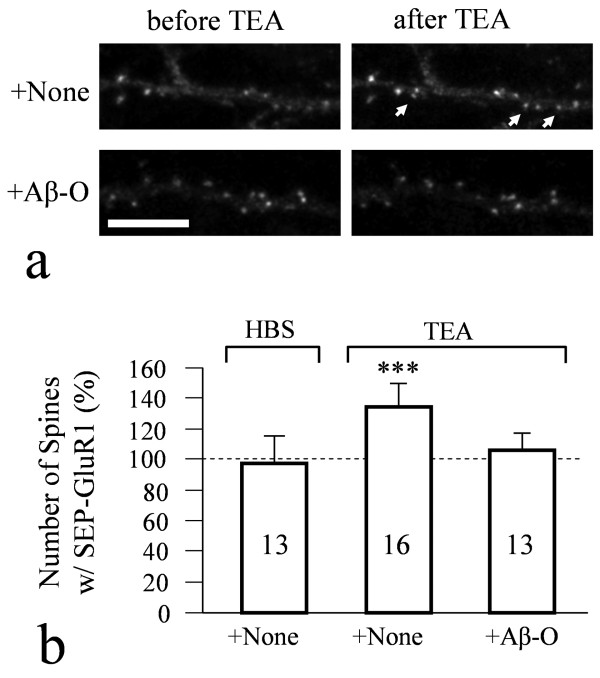
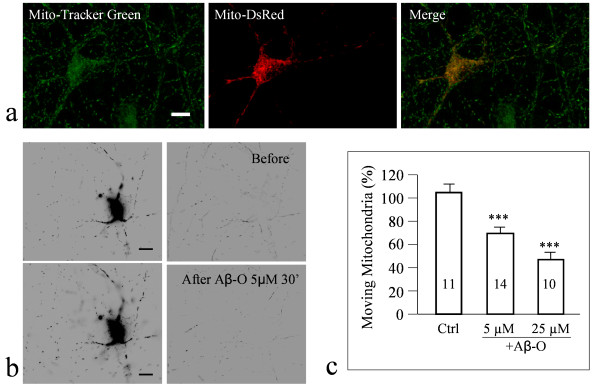
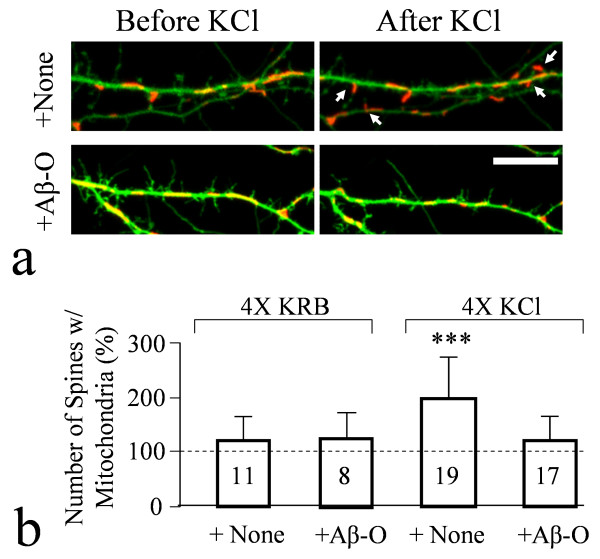
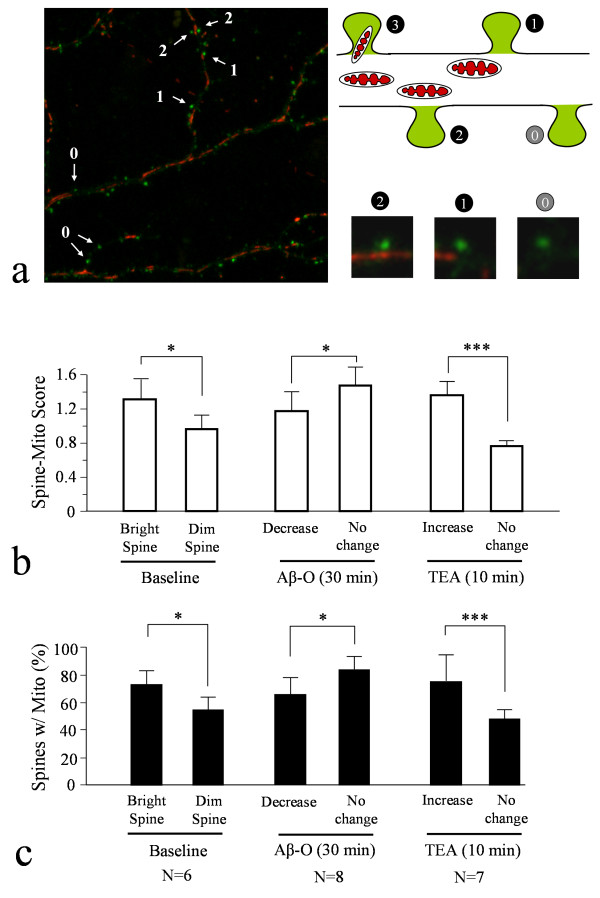
We found that a brief exposure of hippocampal neurons to Abeta oligomers not only led to marked removal of AMPARs from postsynaptic surface but also impaired rapid AMPAR insertion during chemically-induced synaptic potentiation. We also found that Abeta oligomers exerted acute impairment of fast mitochondrial transport, as well as mitochondrial translocation into dendritic spines in response to repetitive membrane depolarization. Quantitative analyses at the single spine level showed a positive correlation between spine-mitochondria association and the surface accumulation of AMPARs. In particular, we found that spines associated with mitochondria tended to be more resistant to Abeta inhibition on AMPAR trafficking. Finally, we showed that inhibition of GSK3beta alleviated Abeta impairment of mitochondrial transport, and effectively abolished Abeta-induced AMPAR loss and inhibition of AMPAR insertion at spines during cLTP.
Our findings indicate that mitochondrial association with dendritic spines may play an important role in supporting AMPAR presence on or trafficking to the postsynaptic membrane. Abeta disruption of mitochondrial trafficking could contribute to AMPAR removal and trafficking defects leading to synaptic inhibition.
突触缺陷是阿尔茨海默病(AD)患者大脑功能改变的主要机制之一。越来越多的研究表明,β-淀粉样蛋白(Abeta)分子的寡聚形式对突触功能有深远的抑制作用,并可能导致突触后表面神经递质受体的大量丢失,但潜在的机制仍知之甚少。在这项研究中,我们研究了线粒体对 Abeta 抑制 AMPA 受体(AMPAR)转运的潜在贡献。
我们发现,海马神经元短暂暴露于 Abeta 寡聚体不仅导致 AMPAR 从突触后表面明显去除,而且还损害了化学诱导的突触增强过程中 AMPAR 的快速插入。我们还发现 Abeta 寡聚体对快速线粒体转运以及线粒体向树突棘的易位产生急性损害,以响应重复的膜去极化。在单个棘突水平的定量分析表明,棘突与线粒体的关联与 AMPAR 表面积累呈正相关。特别是,我们发现与线粒体相关的棘突往往对 Abeta 抑制 AMPAR 转运具有更强的抵抗力。最后,我们表明 GSK3beta 的抑制减轻了 Abeta 对线粒体转运的损害,并有效地消除了 Abeta 在 cLTP 过程中诱导的 AMPAR 丢失和对棘突中 AMPAR 插入的抑制。
我们的发现表明,线粒体与树突棘的关联可能在支持 AMPAR 存在于或转运到突触后膜方面发挥重要作用。Abeta 破坏线粒体转运可能导致 AMPAR 去除和转运缺陷,从而导致突触抑制。