Center of Excellence in Molecular Genetics of Cancer and Human Diseases, Department of Anatomy, Faculty of Medicine, Chulalongkorn University, Bangkok 10330, Thailand.
Mol Cancer. 2010 Mar 31;9:70. doi: 10.1186/1476-4598-9-70.
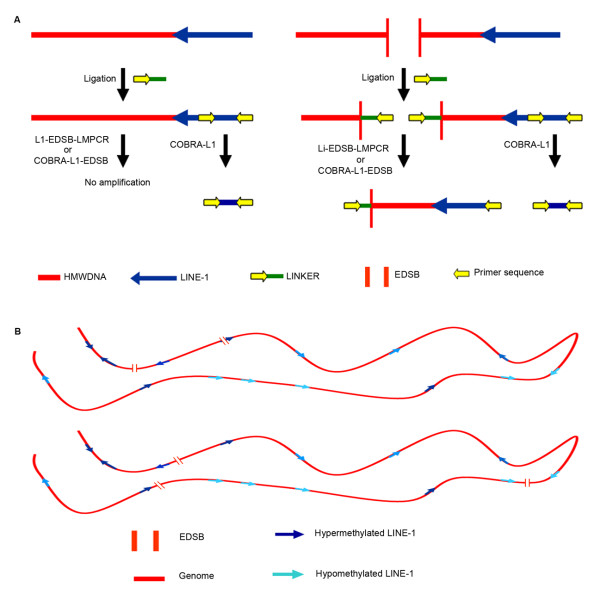
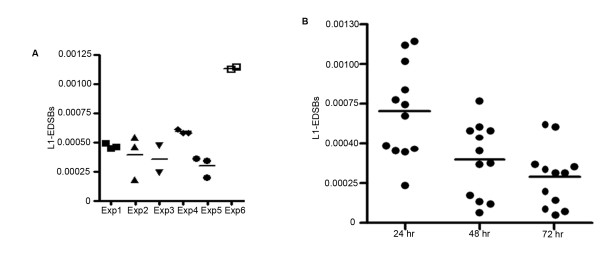
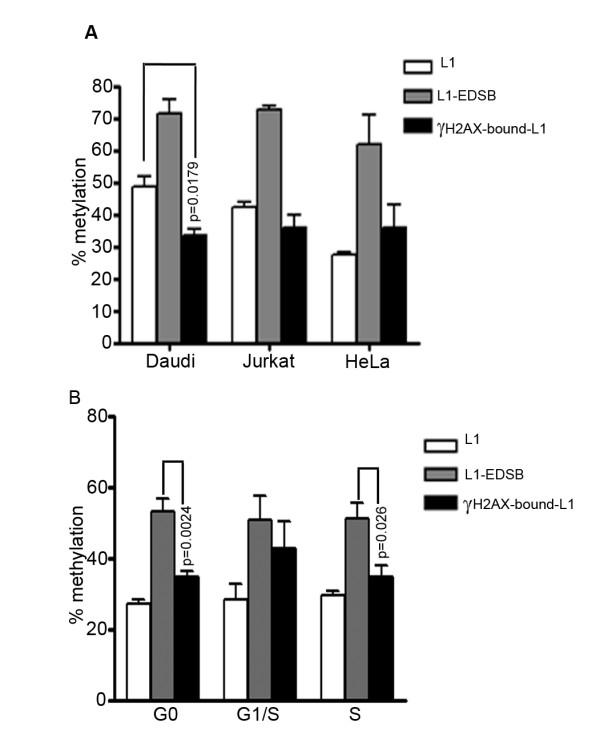
Global hypomethylation and genomic instability are cardinal features of cancers. Recently, we established a method for the detection of DNA methylation levels at sites close to endogenous DNA double strand breaks (EDSBs), and found that those sites have a higher level of methylation than the rest of the genome. Interestingly, the most significant differences between EDSBs and genomes were observed when cells were cultured in the absence of serum. DNA methylation levels on each genomic location are different. Therefore, there are more replication-independent EDSBs (RIND-EDSBs) located in methylated genomic regions. Moreover, methylated and unmethylated RIND-EDSBs are differentially processed. Euchromatins respond rapidly to DSBs induced by irradiation with the phosphorylation of H2AX, gamma-H2AX, and these initiate the DSB repair process. During G0, most DSBs are repaired by non-homologous end-joining repair (NHEJ), mediated by at least two distinct pathways; the Ku-mediated and the ataxia telangiectasia-mutated (ATM)-mediated. The ATM-mediated pathway is more precise. Here we explored how cells process methylated RIND-EDSBs and if RIND-EDSBs play a role in global hypomethylation-induced genomic instability.
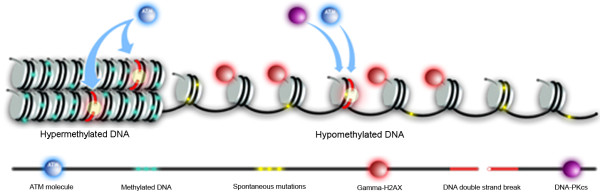
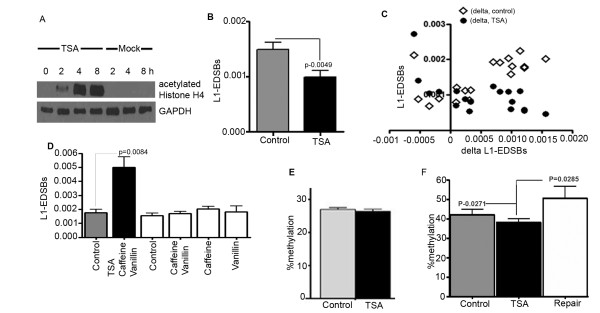
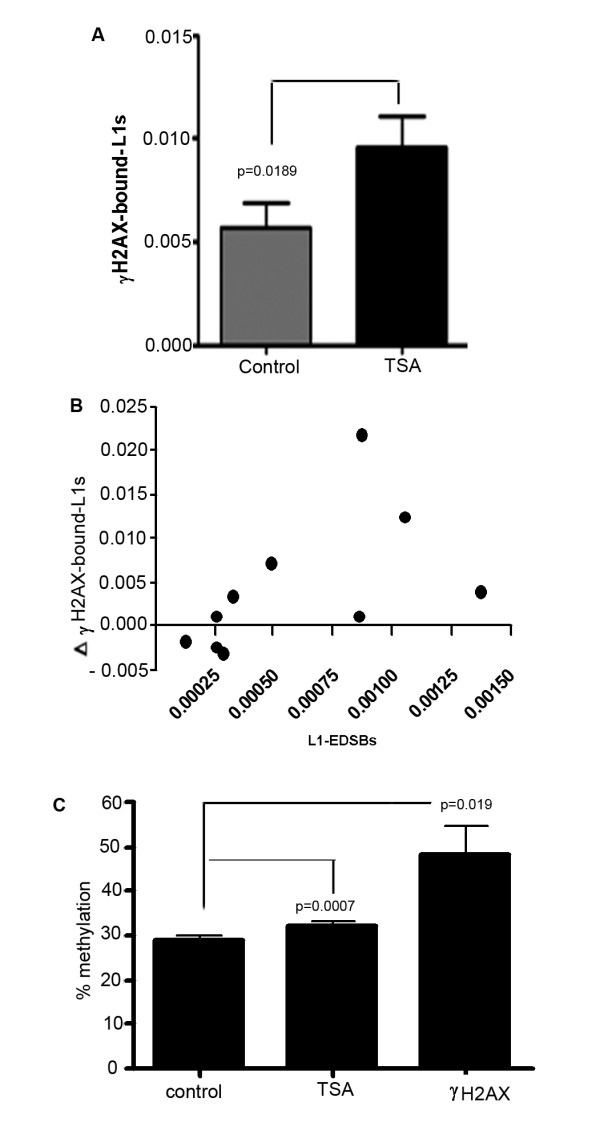
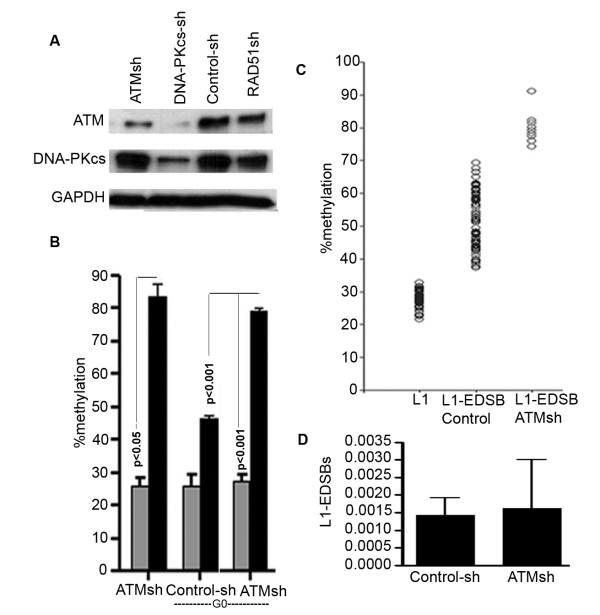
We observed a significant number of methylated RIND-EDSBs that are retained within deacetylated chromatin and free from an immediate cellular response to DSBs, the gamma-H2AX. When cells were treated with tricostatin A (TSA) and the histones became hyperacetylated, the amount of gamma-H2AX-bound DNA increased and the retained RIND-EDSBs were rapidly repaired. When NHEJ was simultaneously inhibited in TSA-treated cells, more EDSBs were detected. Without TSA, a sporadic increase in unmethylated RIND-EDSBs could be observed when Ku-mediated NHEJ was inhibited. Finally, a remarkable increase in RIND-EDSB methylation levels was observed when cells were depleted of ATM, but not of Ku86 and RAD51.
Methylated RIND-EDSBs are retained in non-acetylated heterochromatin because there is a prolonged time lag between RIND-EDSB production and repair. The rapid cellular responses to DSBs may be blocked by compact heterochromatin structure which then allows these breaks to be repaired by a more precise ATM-dependent pathway. In contrast, Ku-mediated NHEJ can repair euchromatin-associated EDSBs. Consequently, spontaneous mutations in hypomethylated genome are produced at faster rates because unmethylated EDSBs are unable to avoid the more error-prone NHEJ mechanisms.
全球低甲基化和基因组不稳定性是癌症的主要特征。最近,我们建立了一种检测接近内源性 DNA 双链断裂(EDSBs)的 DNA 甲基化水平的方法,发现这些位点的甲基化水平高于基因组的其他部分。有趣的是,当细胞在没有血清的情况下培养时,EDSBs 和基因组之间的差异最为显著。每个基因组位置的 DNA 甲基化水平不同。因此,位于甲基化基因组区域的复制独立 EDSBs(RIND-EDSBs)更多。此外,甲基化和非甲基化的 RIND-EDSBs 被不同地处理。常染色质对由辐射引起的 DSBs 反应迅速,通过 H2AX、γ-H2AX 的磷酸化,这些启动 DSB 修复过程。在 G0 期,大多数 DSBs 通过非同源末端连接修复(NHEJ)修复,由至少两种不同途径介导;Ku 介导和共济失调毛细血管扩张突变(ATM)介导。ATM 介导的途径更精确。在这里,我们探讨了细胞如何处理甲基化的 RIND-EDSBs,以及 RIND-EDSBs 是否在全球低甲基化诱导的基因组不稳定性中发挥作用。
我们观察到大量的甲基化 RIND-EDSBs 保留在去乙酰化染色质中,并且没有细胞对 DSB 的立即反应,即γ-H2AX。当细胞用三氯乙酸 A(TSA)处理,组蛋白变得超乙酰化时,结合γ-H2AX 的 DNA 量增加,保留的 RIND-EDSBs 被迅速修复。当 TSA 处理的细胞同时抑制 NHEJ 时,检测到更多的 EDSBs。没有 TSA,当 Ku 介导的 NHEJ 被抑制时,偶尔会观察到未甲基化的 RIND-EDSBs 的增加。最后,当细胞耗尽 ATM 时,RIND-EDSB 甲基化水平显著增加,但 Ku86 和 RAD51 没有耗尽。
甲基化的 RIND-EDSBs 保留在非乙酰化的异染色质中,因为 RIND-EDSB 的产生和修复之间存在时间滞后。DSB 快速的细胞反应可能被紧密的异染色质结构阻断,然后允许这些断裂通过更精确的 ATM 依赖性途径修复。相比之下,Ku 介导的 NHEJ 可以修复常染色质相关的 EDSBs。因此,由于未甲基化的 EDSBs 无法避免更易错的 NHEJ 机制,因此在低甲基化基因组中自发产生突变的速度更快。