School of Computer Science and Informatics, University College Dublin, Dublin, Ireland.
BMC Bioinformatics. 2010 Apr 20;11:197. doi: 10.1186/1471-2105-11-197.
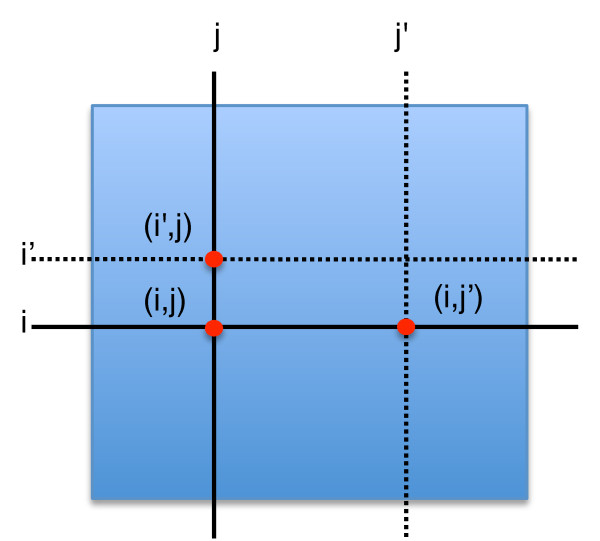
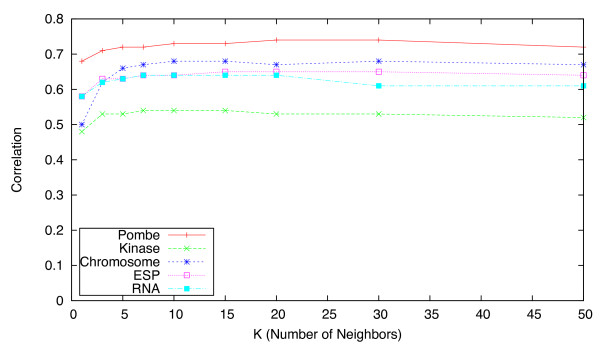
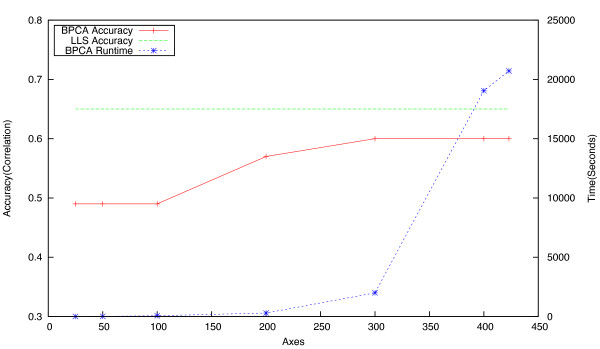
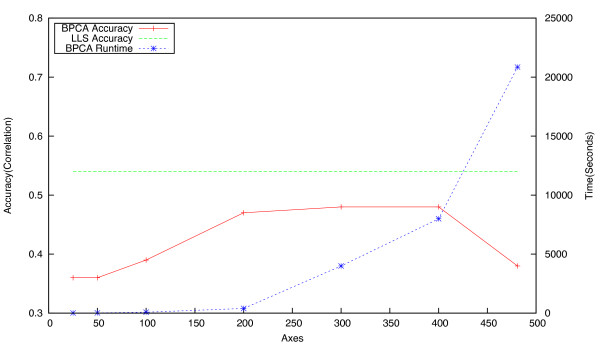
Epistatic miniarray profiling (E-MAPs) is a high-throughput approach capable of quantifying aggravating or alleviating genetic interactions between gene pairs. The datasets resulting from E-MAP experiments typically take the form of a symmetric pairwise matrix of interaction scores. These datasets have a significant number of missing values - up to 35% - that can reduce the effectiveness of some data analysis techniques and prevent the use of others. An effective method for imputing interactions would therefore increase the types of possible analysis, as well as increase the potential to identify novel functional interactions between gene pairs. Several methods have been developed to handle missing values in microarray data, but it is unclear how applicable these methods are to E-MAP data because of their pairwise nature and the significantly larger number of missing values. Here we evaluate four alternative imputation strategies, three local (Nearest neighbor-based) and one global (PCA-based), that have been modified to work with symmetric pairwise data.
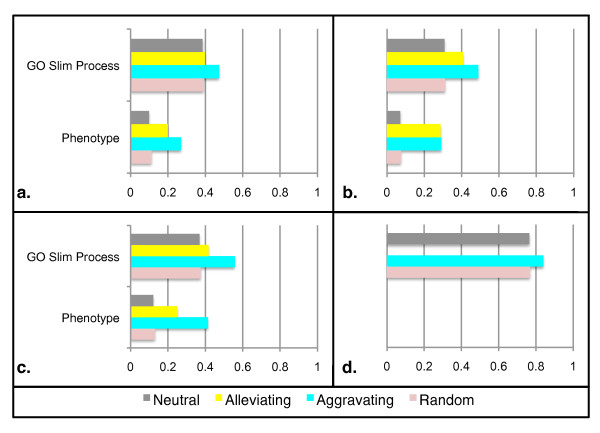
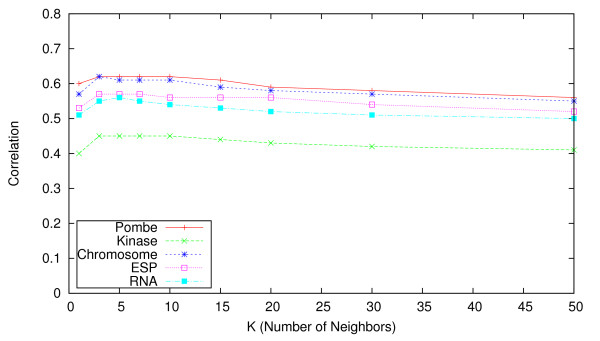
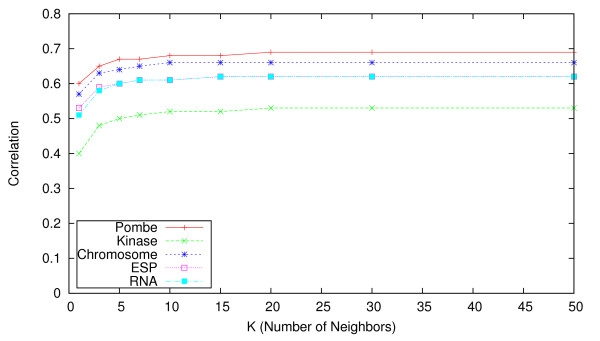
We identify different categories for the missing data based on their underlying cause, and show that values from the largest category can be imputed effectively. We compare local and global imputation approaches across a variety of distinct E-MAP datasets, showing that both are competitive and preferable to filling in with zeros. In addition we show that these methods are effective in an E-MAP from a different species, suggesting that pairwise imputation techniques will be increasingly useful as analogous epistasis mapping techniques are developed in different species. We show that strongly alleviating interactions are significantly more difficult to predict than strongly aggravating interactions. Finally we show that imputed interactions, generated using nearest neighbor methods, are enriched for annotations in the same manner as measured interactions. Therefore our method potentially expands the number of mapped epistatic interactions. In addition we make implementations of our algorithms available for use by other researchers.
We address the problem of missing value imputation for E-MAPs, and suggest the use of symmetric nearest neighbor based approaches as they offer consistently accurate imputations across multiple datasets in a tractable manner.
上位微型数组分析(E-MAPs)是一种高通量方法,能够量化基因对之间的加重或缓解遗传相互作用。E-MAP 实验产生的数据集通常采用交互作用评分的对称成对矩阵形式。这些数据集有大量的缺失值 - 高达 35% - 这可能会降低一些数据分析技术的有效性,并阻止使用其他技术。因此,有效的交互作用插补方法将增加可能的分析类型,并增加识别基因对之间新的功能相互作用的潜力。已经开发了几种处理微阵列数据中缺失值的方法,但由于其成对性质和缺失值数量显著增加,尚不清楚这些方法在 E-MAP 数据中的适用性。在这里,我们评估了四种替代插补策略,三种局部(基于最近邻)和一种全局(基于 PCA),这些方法已被修改为适用于对称成对数据。
我们根据缺失值的潜在原因将其分为不同类别,并表明最大类别的值可以有效插补。我们比较了局部和全局插补方法在各种不同的 E-MAP 数据集上的表现,表明这两种方法都具有竞争力,并且优于用零填充。此外,我们还表明,这些方法在来自不同物种的 E-MAP 中是有效的,这表明随着在不同物种中开发类似的上位映射技术,成对插补技术将越来越有用。我们表明,强烈缓解的相互作用比强烈加重的相互作用更难预测。最后,我们表明,使用最近邻方法生成的插补相互作用与测量相互作用一样,在相同的方式下富集了注释。因此,我们的方法有可能扩展映射的上位相互作用的数量。此外,我们还提供了我们的算法的实现,供其他研究人员使用。
我们解决了 E-MAPs 中缺失值插补的问题,并建议使用对称最近邻方法,因为它们以可管理的方式在多个数据集上提供一致准确的插补。