University of Iowa Carver College of Medicine, Department of Pathology, Iowa City, IA, USA.
Mol Cancer. 2010 Apr 30;9:97. doi: 10.1186/1476-4598-9-97.
Myc is a well known driver of lymphomagenesis, and Myc-activating chromosomal translocation is the recognized hallmark of Burkitt lymphoma, an aggressive form of non-Hodgkin's lymphoma. We developed a model that mimics this translocation event by inserting a mouse Myc cDNA gene into the immunoglobulin heavy chain locus, just upstream of the intronic Emu enhancer. These mice, designated iMyc E mu, readily develop B-cell lymphoma. To study the mechanism of Myc-induced lymphoma, we analyzed signaling pathways in lymphoblastic B-cell lymphomas (LBLs) from iMyc E mu mice, and an LBL-derived cell line, iMyc E mu-1.
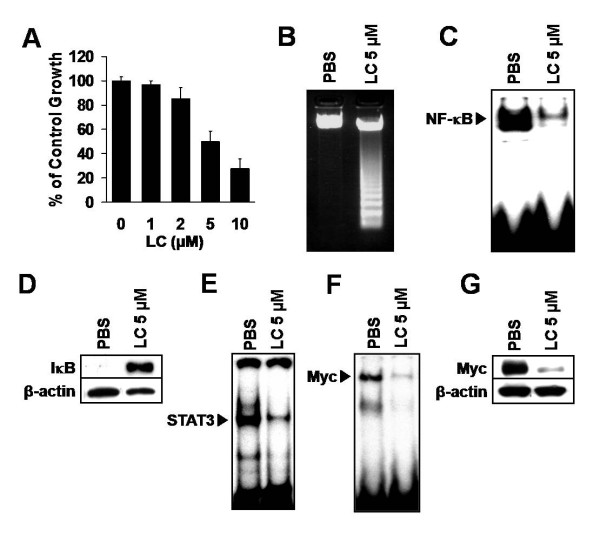
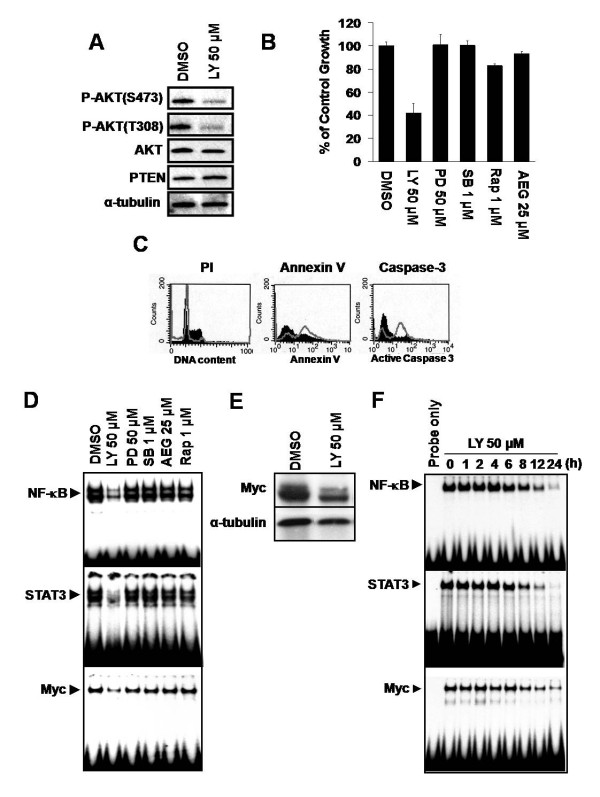
Nuclear factor-kappaB (NF-kappaB) and signal transducer and activator of transcription 3 (STAT3) were constitutively activated in iMyc E mu mice, not only in LBLs but also in the splenic B-lymphocytes of young animals months before tumors developed. Moreover, inhibition of either transcription factor in iMyc E mu-1 cells suppressed growth and caused apoptosis, and the abrogation of NF-kappaB activity reduced DNA binding by both STAT3 and Myc, as well as Myc expression. Inhibition of STAT3 signaling eliminated the activity of both NF-kappaB and Myc, and resulted in a corresponding decrease in the level of Myc. Thus, in iMyc E mu-1 cells NF-kappaB and STAT3 are co-dependent and can both regulate Myc. Consistent with this, NF-kappaB and phosphorylated STAT3 were physically associated with one another. In addition, LBLs and iMyc E mu-1 cells also showed constitutive AKT phosphorylation. Blocking AKT activation by inhibiting PI3K reduced iMyc E mu-1 cell proliferation and caused apoptosis, via downregulation of NF-kappaB and STAT3 activity and a reduction of Myc levels. Co-treatment with NF-kappaB, STAT3 or/and PI3K inhibitors led to additive inhibition of iMyc E mu-1 cell proliferation, suggesting that these signaling pathways converge.
Our findings support the notion that constitutive activation of NF-kappaB and STAT3 depends on upstream signaling through PI3K, and that this activation is important for cell survival and proliferation, as well as for maintaining the level of Myc. Together, these data implicate crosstalk among NF-kappaB, STAT3 and PI3K in the development of iMyc E mu B-cell lymphomas.
Myc 是淋巴癌发生的一个众所周知的驱动因素,而 Myc 激活染色体易位是伯基特淋巴瘤(一种侵袭性非霍奇金淋巴瘤)的公认标志。我们通过将小鼠 Myc cDNA 基因插入免疫球蛋白重链基因座,就在内含子 Emu 增强子的上游,构建了一种模拟这种易位事件的模型。这些被命名为 iMyc E mu 的小鼠容易发生 B 细胞淋巴瘤。为了研究 Myc 诱导的淋巴瘤的机制,我们分析了来自 iMyc E mu 小鼠的淋巴母细胞性 B 细胞淋巴瘤(LBL)和源自 LBL 的细胞系 iMyc E mu-1 的信号通路。
核因子-κB(NF-κB)和信号转导和转录激活因子 3(STAT3)在 iMyc E mu 小鼠中持续激活,不仅在 LBL 中,而且在肿瘤发生前数月的年轻动物的脾脏 B 淋巴细胞中也是如此。此外,在 iMyc E mu-1 细胞中抑制任一转录因子都会抑制细胞生长并导致细胞凋亡,而 NF-κB 活性的阻断会降低 STAT3 和 Myc 的 DNA 结合以及 Myc 的表达。抑制 STAT3 信号通路会消除 NF-κB 和 Myc 的活性,并相应降低 Myc 的水平。因此,在 iMyc E mu-1 细胞中,NF-κB 和 STAT3 是相互依赖的,可以共同调节 Myc。这与 NF-κB 和磷酸化 STAT3 彼此之间存在物理关联的情况一致。此外,LBL 和 iMyc E mu-1 细胞也表现出 AKT 的持续磷酸化。通过抑制 PI3K 阻断 AKT 激活会降低 iMyc E mu-1 细胞的增殖并通过降低 NF-κB 和 STAT3 活性以及降低 Myc 水平导致细胞凋亡。NF-κB、STAT3 和/或 PI3K 抑制剂的联合治疗导致 iMyc E mu-1 细胞增殖的相加抑制,表明这些信号通路汇聚。
我们的研究结果支持这样一种观点,即 NF-κB 和 STAT3 的持续激活依赖于 PI3K 的上游信号转导,并且这种激活对于细胞存活和增殖以及维持 Myc 水平是重要的。综上所述,这些数据表明 NF-κB、STAT3 和 PI3K 之间的相互作用参与了 iMyc E mu B 细胞淋巴瘤的发生。