Department of Statistics, Stanford University, Stanford, California 94305-4065, USA.
BMC Biol. 2010 May 11;8:58. doi: 10.1186/1741-7007-8-58.
Ultra-high throughput sequencing technologies provide opportunities both for discovery of novel molecular species and for detailed comparisons of gene expression patterns. Small RNA populations are particularly well suited to this analysis, as many different small RNAs can be completely sequenced in a single instrument run.
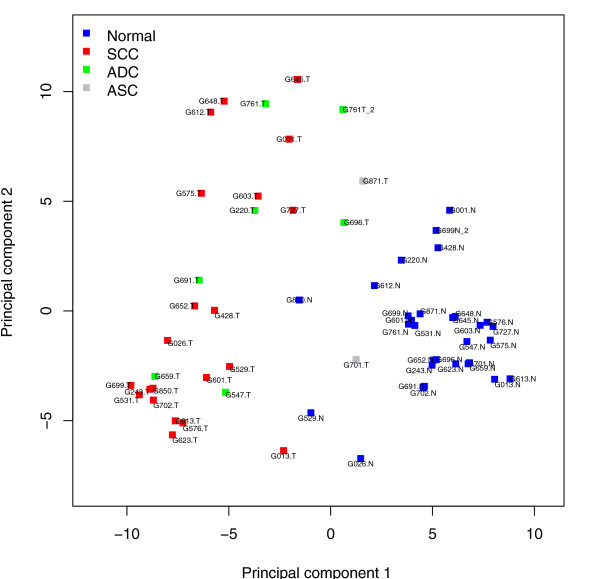
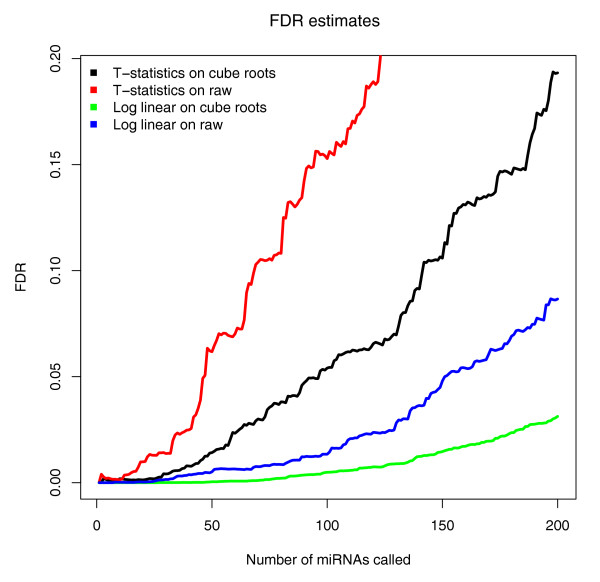
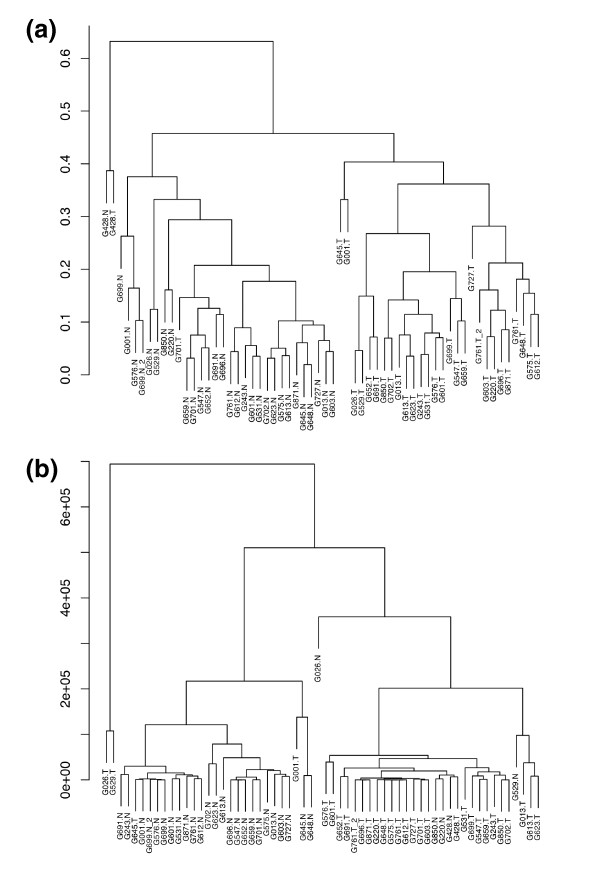
We prepared small RNA libraries from 29 tumour/normal pairs of human cervical tissue samples. Analysis of the resulting sequences (42 million in total) defined 64 new human microRNA (miRNA) genes. Both arms of the hairpin precursor were observed in twenty-three of the newly identified miRNA candidates. We tested several computational approaches for the analysis of class differences between high throughput sequencing datasets and describe a novel application of a log linear model that has provided the most effective analysis for this data. This method resulted in the identification of 67 miRNAs that were differentially-expressed between the tumour and normal samples at a false discovery rate less than 0.001.
This approach can potentially be applied to any kind of RNA sequencing data for analysing differential sequence representation between biological sample sets.
超高通量测序技术为发现新的分子物种和详细比较基因表达模式提供了机会。小 RNA 群体特别适合这种分析,因为在单个仪器运行中可以完全测序许多不同的小 RNA。
我们从 29 对人宫颈组织样本的肿瘤/正常对中制备了小 RNA 文库。对所得序列(总计 4200 万条)的分析定义了 64 个新的人类 microRNA(miRNA)基因。在二十三个新鉴定的 miRNA 候选物中观察到发夹前体的两个臂。我们测试了几种计算方法来分析高通量测序数据集之间的类别差异,并描述了对数线性模型的一种新应用,该模型为该数据提供了最有效的分析。该方法确定了 67 个 miRNA 在肿瘤和正常样本之间的差异表达,假发现率小于 0.001。
这种方法可以应用于任何类型的 RNA 测序数据,用于分析生物样本组之间的差异序列表示。