School of Information Technology, Jawaharlal Nehru University, New Delhi, India.
BMC Genomics. 2010 May 7;11:288. doi: 10.1186/1471-2164-11-288.
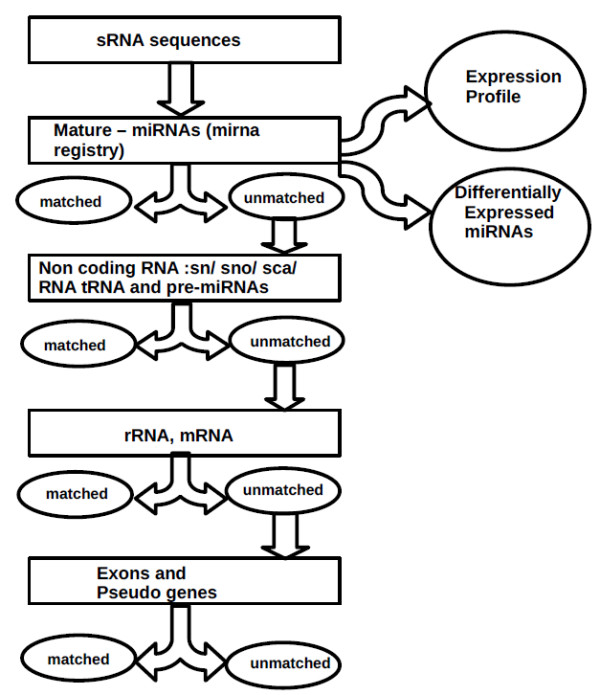
MicroRNAs are a class of small non-coding RNAs that regulate mRNA expression at the post - transcriptional level and thereby many fundamental biological processes. A number of methods, such as multiplex polymerase chain reaction, microarrays have been developed for profiling levels of known miRNAs. These methods lack the ability to identify novel miRNAs and accurately determine expression at a range of concentrations. Deep or massively parallel sequencing methods are providing suitable platforms for genome wide transcriptome analysis and have the ability to identify novel transcripts.
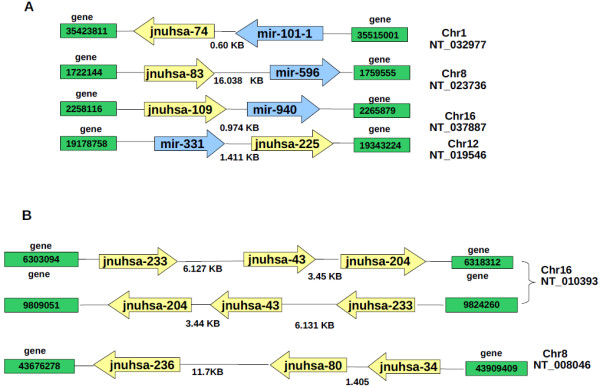
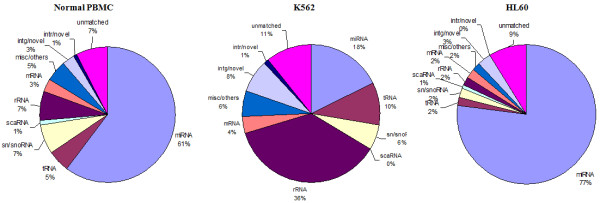
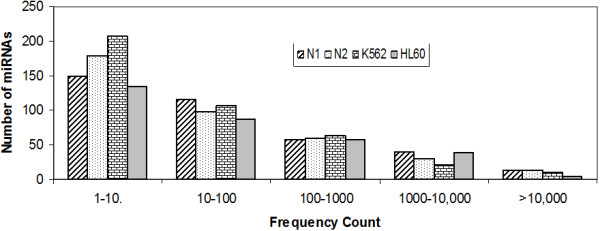
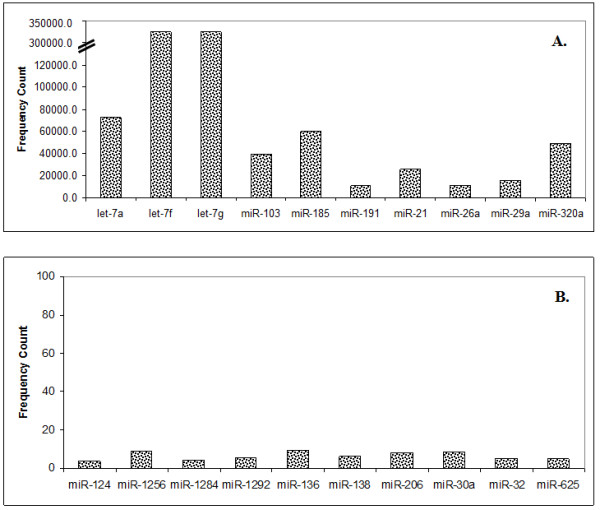
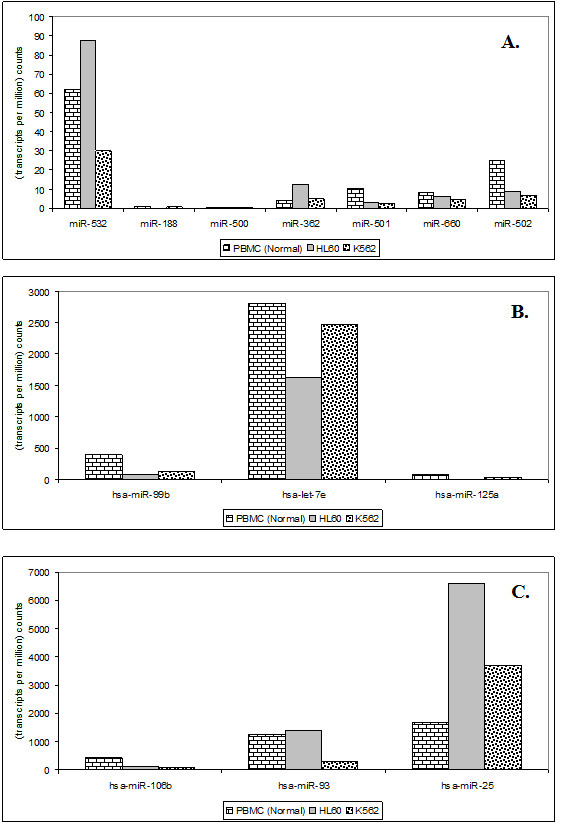
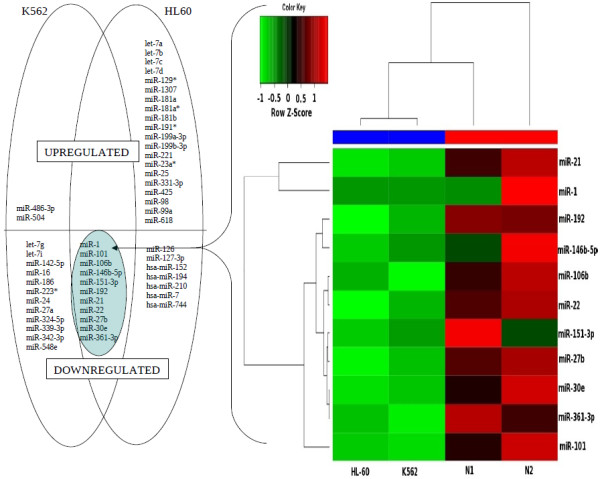
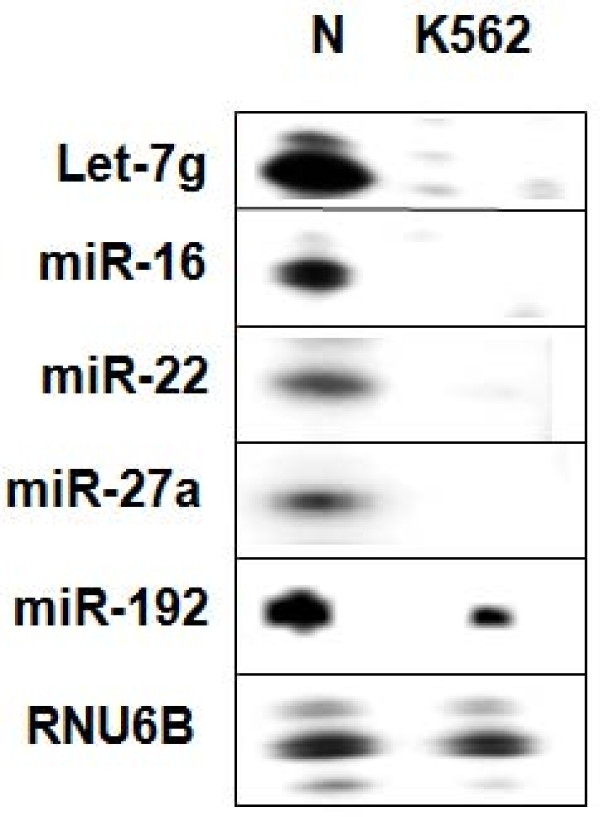
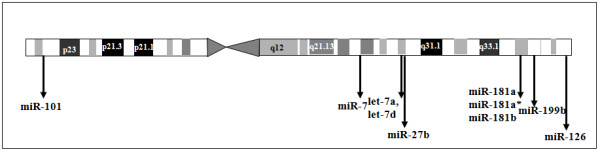
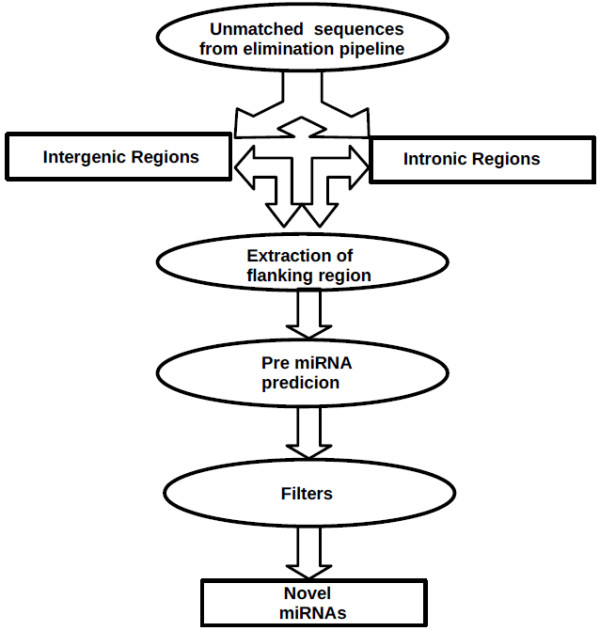
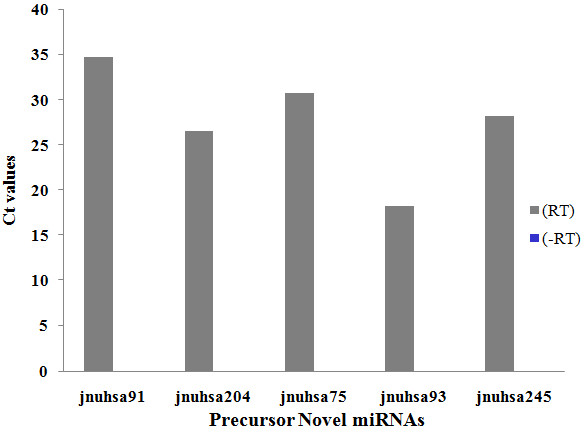
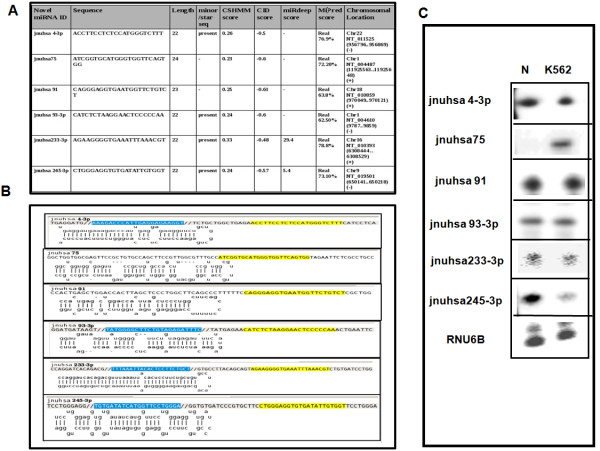
The results of analysis of small RNA sequences obtained by Solexa technology of normal peripheral blood mononuclear cells, tumor cell lines K562 and HL60 are presented. In general K562 cells displayed overall low level of miRNA population and also low levels of DICER. Some of the highly expressed miRNAs in the leukocytes include several members of the let-7 family, miR-21, 103, 185, 191 and 320a. Comparison of the miRNA profiles of normal versus K562 or HL60 cells revealed a specific set of differentially expressed molecules. Correlation of the miRNA with that of mRNA expression profiles, obtained by microarray, revealed a set of target genes showing inverse correlation with miRNA levels. Relative expression levels of individual miRNAs belonging to a cluster were found to be highly variable. Our computational pipeline also predicted a number of novel miRNAs. Some of the predictions were validated by Real-time RT-PCR and or RNase protection assay. Organization of some of the novel miRNAs in human genome suggests that these may also be part of existing clusters or form new clusters.
We conclude that about 904 miRNAs are expressed in human leukocytes. Out of these 370 are novel miRNAs. We have identified miRNAs that are differentially regulated in normal PBMC with respect to cancer cells, K562 and HL60. Our results suggest that post - transcriptional processes may play a significant role in regulating levels of miRNAs in tumor cells. The study also provides a customized automated computation pipeline for miRNA profiling and identification of novel miRNAs; even those that are missed out by other existing pipelines. The Computational Pipeline is available at the website: http://mirna.jnu.ac.in/deep_sequencing/deep_sequencing.html.
miRNAs 是一类小的非编码 RNA,通过在转录后水平调节 mRNA 的表达,从而调控许多基本的生物过程。已经开发了多种方法来分析已知 miRNAs 的水平,例如多重聚合酶链反应、微阵列。这些方法缺乏鉴定新 miRNAs 的能力,并且无法准确地在一系列浓度下测定表达水平。深度测序或大规模平行测序方法为全基因组转录组分析提供了合适的平台,并且具有鉴定新转录本的能力。
本文展示了使用 Solexa 技术对正常外周血单个核细胞、肿瘤细胞系 K562 和 HL60 进行小 RNA 序列分析的结果。总体而言,K562 细胞显示 miRNA 群体的总体水平较低,并且 DICER 的水平也较低。白细胞中高度表达的 miRNAs 包括 let-7 家族的几个成员、miR-21、103、185、191 和 320a。正常细胞与 K562 或 HL60 细胞的 miRNA 谱比较显示出一组特定的差异表达分子。通过微阵列获得的 miRNA 与 mRNA 表达谱的相关性分析显示,一组靶基因与 miRNA 水平呈负相关。属于簇的单个 miRNAs 的相对表达水平发现具有高度可变性。我们的计算管道还预测了一些新的 miRNAs。其中一些预测通过实时 RT-PCR 和或 RNase 保护测定得到验证。一些新 miRNAs 在人类基因组中的组织表明,它们也可能是现有簇的一部分或形成新的簇。
我们得出结论,约有 904 个 miRNAs 在人类白细胞中表达。其中 370 个是新的 miRNAs。我们已经鉴定出在正常 PBMC 与癌细胞 K562 和 HL60 相比差异表达的 miRNAs。我们的结果表明,转录后过程可能在调节肿瘤细胞中 miRNAs 的水平方面发挥重要作用。该研究还提供了用于 miRNA 分析和鉴定新 miRNAs 的定制自动化计算管道;即使是其他现有管道错过的 miRNAs 也可以鉴定出来。计算管道可在以下网站获得:http://mirna.jnu.ac.in/deep_sequencing/deep_sequencing.html。