Science for Life Laboratory, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden.
Bioinformatics. 2010 Jul 1;26(13):1595-600. doi: 10.1093/bioinformatics/btq230. Epub 2010 May 13.
New generation sequencing technologies producing increasingly complex datasets demand new efficient and specialized sequence analysis algorithms. Often, it is only the 'novel' sequences in a complex dataset that are of interest and the superfluous sequences need to be removed.
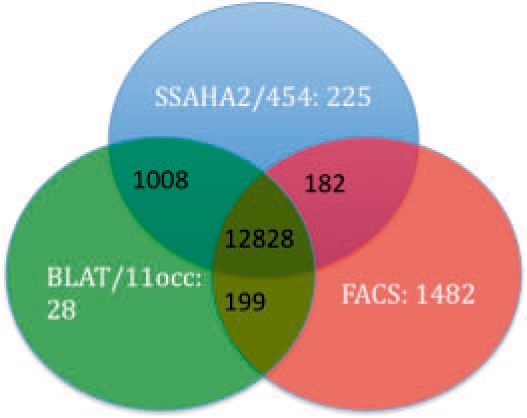
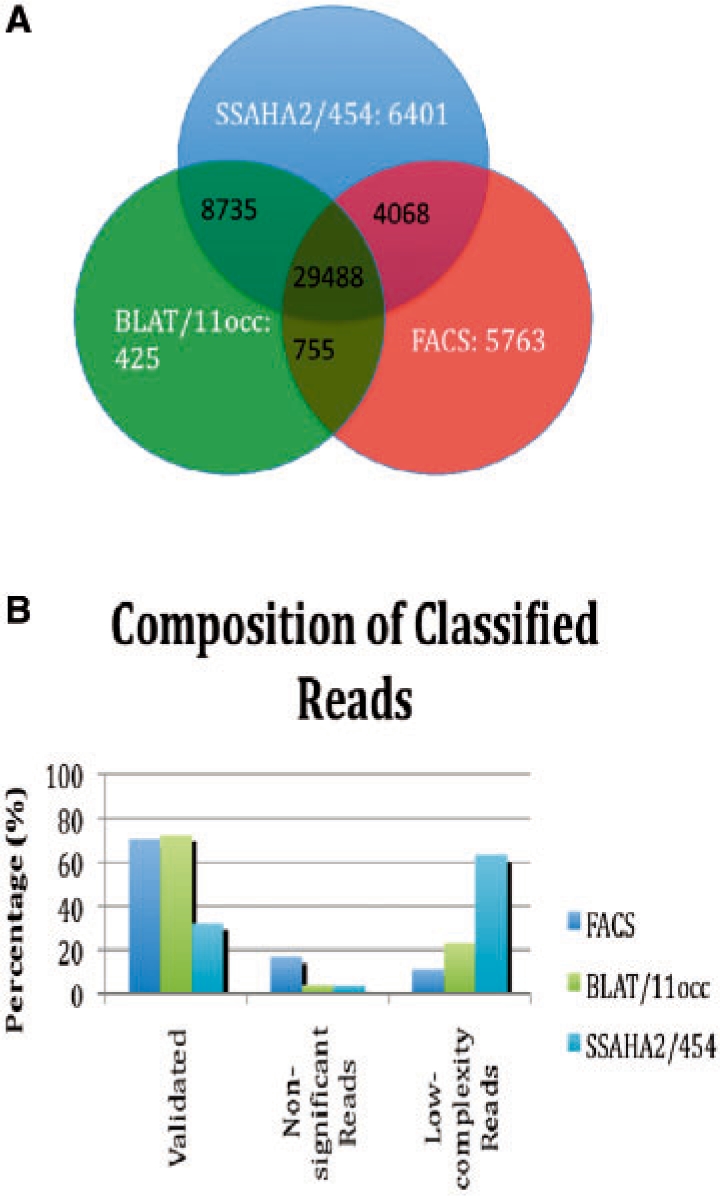
A novel algorithm, fast and accurate classification of sequences (FACSs), is introduced that can accurately and rapidly classify sequences as belonging or not belonging to a reference sequence. FACS was first optimized and validated using a synthetic metagenome dataset. An experimental metagenome dataset was then used to show that FACS achieves comparable accuracy as BLAT and SSAHA2 but is at least 21 times faster in classifying sequences.
Source code for FACS, Bloom filters and MetaSim dataset used is available at http://facs.biotech.kth.se. The Bloom::Faster 1.6 Perl module can be downloaded from CPAN at http://search.cpan.org/ approximately palvaro/Bloom-Faster-1.6/
henrik.stranneheim@biotech.kth.se; joakiml@biotech.kth.se
Supplementary data are available at Bioinformatics online.
新一代测序技术产生的日益复杂的数据集需要新的高效和专业的序列分析算法。通常,只有复杂数据集中的“新颖”序列才是感兴趣的,而多余的序列需要被去除。
引入了一种新颖的算法,快速准确的序列分类(FACS),可以准确快速地将序列分类为属于或不属于参考序列。FACS 首先使用合成宏基因组数据集进行了优化和验证。然后使用实验宏基因组数据集表明,FACS 实现了与 BLAT 和 SSAHA2 相当的准确性,但在分类序列时至少快 21 倍。
FACS、Bloom 过滤器和 MetaSim 数据集的源代码可在 http://facs.biotech.kth.se 获得。Bloom::Faster 1.6 Perl 模块可从 CPAN 下载,网址为 http://search.cpan.org/approximately palvaro/Bloom-Faster-1.6/
henrik.stranneheim@biotech.kth.se;joakiml@biotech.kth.se
补充数据可在 Bioinformatics 在线获得。