Katebi Ataur R, Kloczkowski Andrzej, Jernigan Robert L
L.H. Baker Center for Bioinformatics and Biological Statistics, Iowa State University, Ames, Iowa 50011-0320, USA.
BMC Struct Biol. 2010 May 17;10 Suppl 1(Suppl 1):S4. doi: 10.1186/1472-6807-10-S1-S4.
Currently a huge amount of protein-protein interaction data is available from high throughput experimental methods. In a large network of protein-protein interactions, groups of proteins can be identified as functional clusters having related functions where a single protein can occur in multiple clusters. However experimental methods are error-prone and thus the interactions in a functional cluster may include false positives or there may be unreported interactions. Therefore correctly identifying a functional cluster of proteins requires the knowledge of whether any two proteins in a cluster interact, whether an interaction can exclude other interactions, or how strong the affinity between two interacting proteins is.
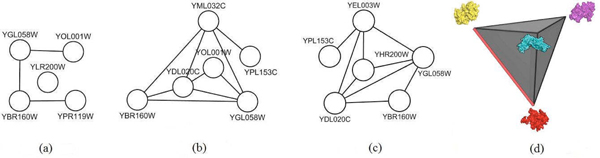
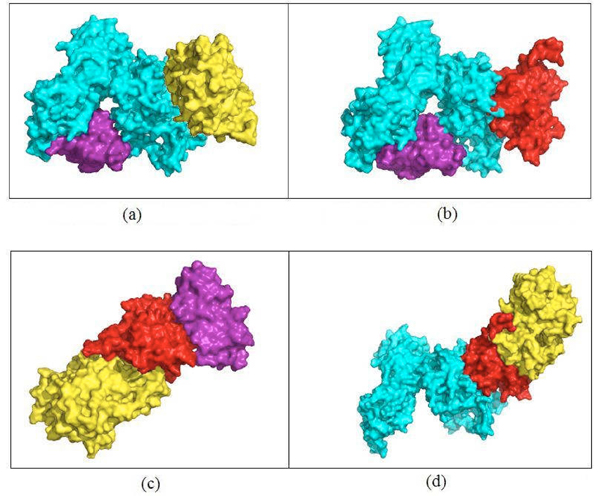
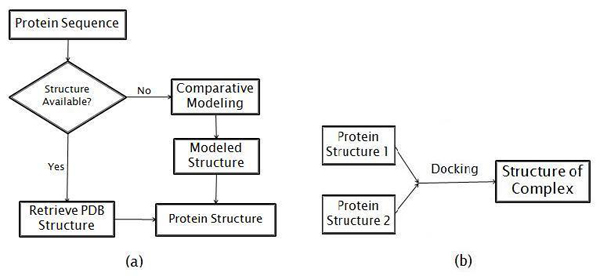
In the present work the yeast protein-protein interaction network is clustered using a spectral clustering method proposed by us in 2006 and the individual clusters are investigated for functional relationships among the member proteins. 3D structural models of the proteins in one cluster have been built--the protein structures are retrieved from the Protein Data Bank or predicted using a comparative modeling approach. A rigid body protein docking method (Cluspro) is used to predict the protein-protein interaction complexes. Binding sites of the docked complexes are characterized by their buried surface areas in the docked complexes, as a measure of the strength of an interaction.
The clustering method yields functionally coherent clusters. Some of the interactions in a cluster exclude other interactions because of shared binding sites. New interactions among the interacting proteins are uncovered, and thus higher order protein complexes in the cluster are proposed. Also the relative stability of each of the protein complexes in the cluster is reported.
Although the methods used are computationally expensive and require human intervention and judgment, they can identify the interactions that could occur together or ones that are mutually exclusive. In addition indirect interactions through another intermediate protein can be identified. These theoretical predictions might be useful for crystallographers to select targets for the X-ray crystallographic determination of protein complexes.
目前,通过高通量实验方法可获得大量蛋白质-蛋白质相互作用数据。在一个庞大的蛋白质-蛋白质相互作用网络中,蛋白质组可被识别为具有相关功能的功能簇,其中单个蛋白质可能出现在多个簇中。然而,实验方法容易出错,因此功能簇中的相互作用可能包括假阳性,或者可能存在未报告的相互作用。因此,正确识别蛋白质功能簇需要了解簇中任意两个蛋白质是否相互作用、一种相互作用是否能排除其他相互作用,或者两个相互作用蛋白质之间的亲和力有多强。
在本研究中,使用我们在2006年提出的光谱聚类方法对酵母蛋白质-蛋白质相互作用网络进行聚类,并研究各个簇中成员蛋白质之间的功能关系。构建了一个簇中蛋白质的三维结构模型——从蛋白质数据库中检索蛋白质结构或使用比较建模方法进行预测。使用刚体蛋白质对接方法(Cluspro)预测蛋白质-蛋白质相互作用复合物。对接复合物的结合位点通过其在对接复合物中的埋藏表面积来表征,作为相互作用强度的一种度量。
聚类方法产生功能上连贯的簇。簇中的一些相互作用由于共享结合位点而排除其他相互作用。发现了相互作用蛋白质之间的新相互作用,因此提出了簇中更高阶的蛋白质复合物。还报告了簇中每个蛋白质复合物的相对稳定性。
虽然所使用的方法计算成本高,需要人工干预和判断,但它们可以识别可能一起发生的相互作用或相互排斥的相互作用。此外,可以识别通过另一种中间蛋白质的间接相互作用。这些理论预测可能有助于晶体学家选择用于蛋白质复合物X射线晶体学测定的靶标。