Department of Epidemiology and Biostatistics, School of Public Health, Imperial College, St, Mary's Campus, Norfolk Place London W2 1PG, UK.
BMC Bioinformatics. 2010 May 20;11:270. doi: 10.1186/1471-2105-11-270.
In microarray studies researchers are often interested in the comparison of relevant quantities between two or more similar experiments, involving different treatments, tissues, or species. Typically each experiment reports measures of significance (e.g. p-values) or other measures that rank its features (e.g genes). Our objective is to find a list of features that are significant in all experiments, to be further investigated. In this paper we present an R package called sdef, that allows the user to quantify the evidence of communality between the experiments using previously proposed statistical methods based on the ranked lists of p-values. sdef implements two approaches that address this objective: the first is a permutation test of the maximal ratio of observed to expected common features under the hypothesis of independence between the experiments. The second approach, set in a Bayesian framework, is more flexible as it takes into account the uncertainty on the number of genes differentially expressed in each experiment.
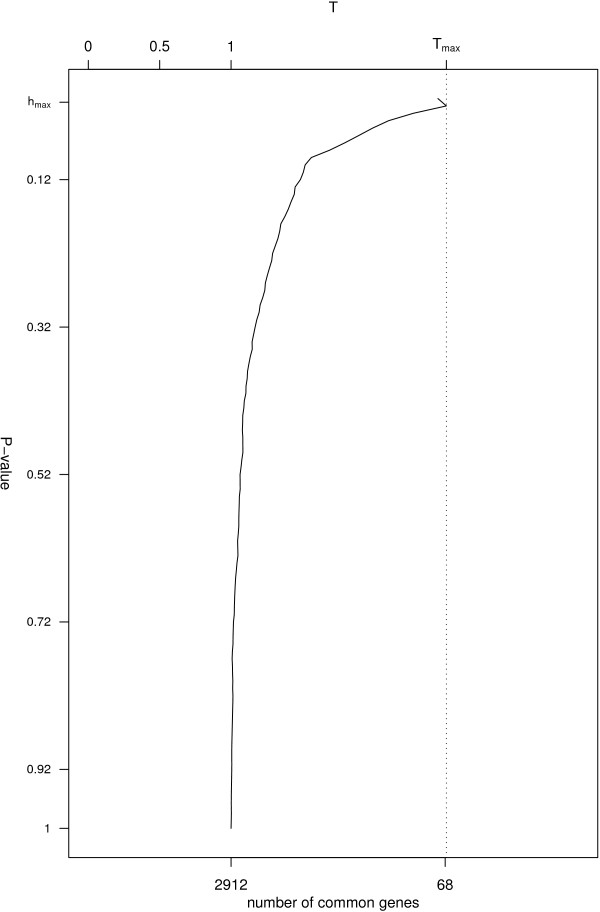
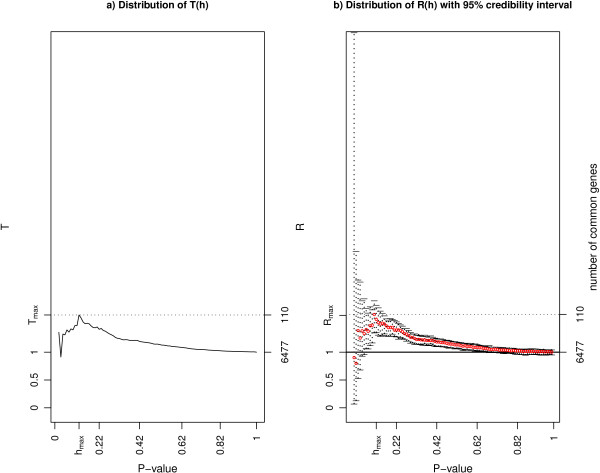
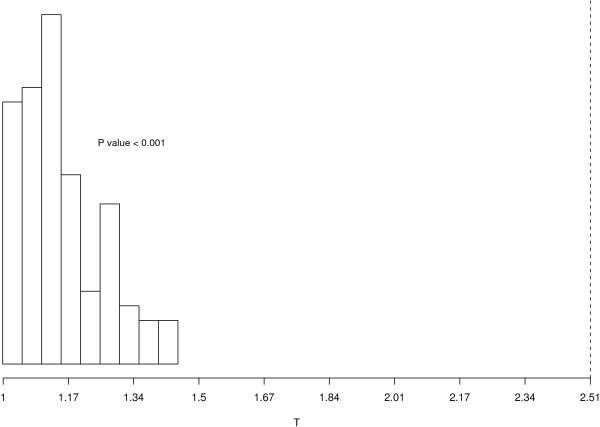
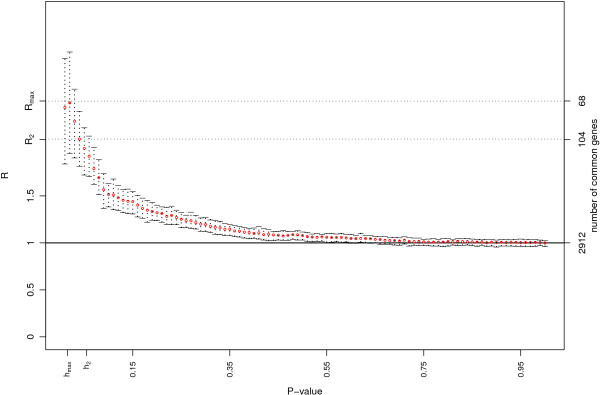
We used sdef to re-analyze publicly available data i) on Type 2 diabetes susceptibility in mice on liver and skeletal muscle (two experiments); ii) on molecular similarities between mammalian sexes (three experiments). For the first example, we found between 68 and 104 genes commonly perturbed between the two tissues, using the two methods described above, and enrichment of the inflammation pathways, which are related to obesity and diabetes. For the second example, looking at three lists of features, we found 110 genes commonly perturbed between the three tissues, using the same two methods, and enrichment on genes involved in cell development.
sdef is an R package that provides researchers with an easy and powerful methodology to find lists of features commonly perturbed in two or more experiments to be further investigated. The package is provided with plots and tables to help the user visualize and interpret the results. The Windows, Linux and MacOS versions of the package, together with the documentation are available on the website http://cran.r-project.org/web/packages/sdef/index.html.
在微阵列研究中,研究人员通常有兴趣比较两个或更多相似实验之间的相关数量,这些实验涉及不同的处理、组织或物种。每个实验通常会报告度量的显著性(例如 p 值)或其他度量标准,这些度量标准会对其特征(例如基因)进行排名。我们的目标是找到在所有实验中都显著的特征列表,以便进一步研究。本文介绍了一个名为 sdef 的 R 包,该包允许用户使用基于 p 值排名列表的先前提出的统计方法来量化实验之间的共性证据。sdef 实现了两种方法来解决这个问题:第一种方法是在实验之间独立的假设下,对观察到的共同特征与预期共同特征的最大比值进行置换检验。第二种方法是在贝叶斯框架下,它更加灵活,因为它考虑了每个实验中差异表达基因数量的不确定性。
我们使用 sdef 重新分析了公开可用的数据,i)在肝脏和骨骼肌中的 2 型糖尿病易感性的小鼠(两个实验);ii)哺乳动物性别之间的分子相似性(三个实验)。对于第一个例子,我们使用上述两种方法在两个组织之间找到了 68 到 104 个共同受干扰的基因,并发现了与肥胖和糖尿病相关的炎症途径的富集。对于第二个例子,我们查看了三个特征列表,使用相同的两种方法在三个组织之间找到了 110 个共同受干扰的基因,并发现了与细胞发育相关的基因的富集。
sdef 是一个 R 包,为研究人员提供了一种简单而强大的方法来找到在两个或更多实验中普遍受到干扰的特征列表,以便进一步研究。该包提供了图表和表格,以帮助用户可视化和解释结果。该包的 Windows、Linux 和 MacOS 版本以及文档可在网站 http://cran.r-project.org/web/packages/sdef/index.html 上获得。