Department of Epidemiology and Biostatistics, Memorial Sloan-Kettering Cancer Center, New York, NY, USA.
BMC Bioinformatics. 2010 Jun 2;11:297. doi: 10.1186/1471-2105-11-297.
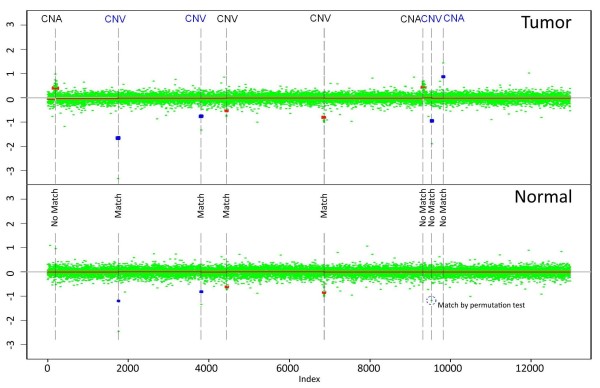
Both somatic copy number alterations (CNAs) and germline copy number variants (CNVs) that are prevalent in healthy individuals can appear as recurrent changes in comparative genomic hybridization (CGH) analyses of tumors. In order to identify important cancer genes CNAs and CNVs must be distinguished. Although the Database of Genomic Variants (DGV) contains a list of all known CNVs, there is no standard methodology to use the database effectively.
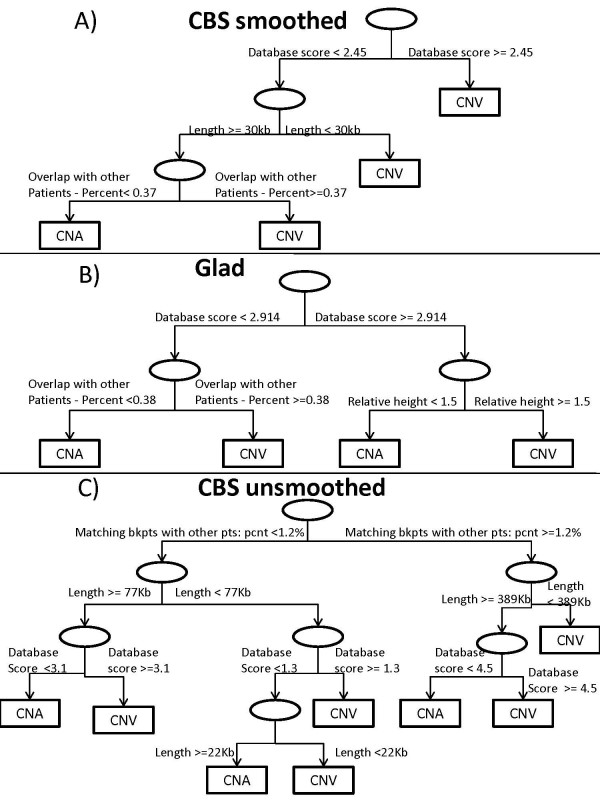
We develop a prediction model that distinguishes CNVs from CNAs based on the information contained in the DGV and several other variables, including segment's length, height, closeness to a telomere or centromere and occurrence in other patients. The models are fitted on data from glioblastoma and their corresponding normal samples that were collected as part of The Cancer Genome Atlas project and hybridized to Agilent 244 K arrays.
Using the DGV alone CNVs in the test set can be correctly identified with about 85% accuracy if the outliers are removed before segmentation and with 72% accuracy if the outliers are included, and additional variables improve the prediction by about 2-3% and 12%, respectively. Final models applied to data from ovarian tumors have about 90% accuracy with all the variables and 86% accuracy with the DGV alone.
在肿瘤比较基因组杂交(CGH)分析中,常见于健康个体的体细胞拷贝数改变(CNAs)和种系拷贝数变异(CNVs)都可能表现为反复出现的变化。为了识别重要的癌症基因,必须区分 CNA 和 CNV。尽管基因组变异数据库(DGV)包含所有已知 CNV 的列表,但目前还没有有效的标准方法来使用该数据库。
我们开发了一种基于 DGV 和其他几个变量(包括片段长度、高度、与端粒或着丝粒的接近程度以及在其他患者中的发生情况)的预测模型,用于区分 CNV 和 CNA。该模型基于癌症基因组图谱项目中收集的胶质母细胞瘤及其相应正常样本的杂交到 Agilent 244K 芯片上的数据进行拟合。
如果在分割前先移除异常值,仅使用 DGV 可以将测试集中的 CNV 正确识别,准确率约为 85%;如果包含异常值,准确率约为 72%。额外的变量分别提高了约 2-3%和 12%的预测准确性。最终应用于卵巢肿瘤数据的模型在包含所有变量时准确率约为 90%,仅使用 DGV 时准确率约为 86%。