Scripps Genomic Medicine, Scripps Translational Science Institute, La Jolla, CA 92037, USA.
Bioinformatics. 2010 Jun 15;26(12):i318-24. doi: 10.1093/bioinformatics/btq214.
Next-generation sequencing technologies have enabled the sequencing of several human genomes in their entirety. However, the routine resequencing of complete genomes remains infeasible. The massive capacity of next-generation sequencers can be harnessed for sequencing specific genomic regions in hundreds to thousands of individuals. Sequencing-based association studies are currently limited by the low level of multiplexing offered by sequencing platforms. Pooled sequencing represents a cost-effective approach for studying rare variants in large populations. To utilize the power of DNA pooling, it is important to accurately identify sequence variants from pooled sequencing data. Detection of rare variants from pooled sequencing represents a different challenge than detection of variants from individual sequencing.
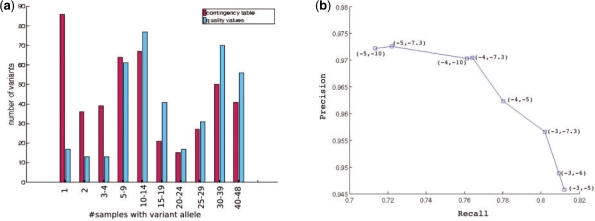
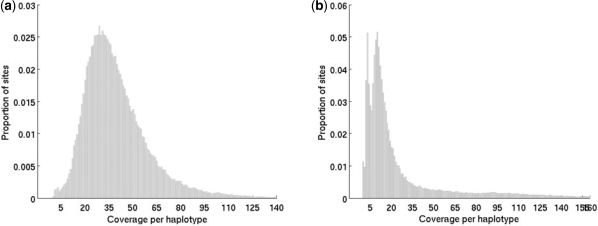
We describe a novel statistical approach, CRISP [Comprehensive Read analysis for Identification of Single Nucleotide Polymorphisms (SNPs) from Pooled sequencing] that is able to identify both rare and common variants by using two approaches: (i) comparing the distribution of allele counts across multiple pools using contingency tables and (ii) evaluating the probability of observing multiple non-reference base calls due to sequencing errors alone. Information about the distribution of reads between the forward and reverse strands and the size of the pools is also incorporated within this framework to filter out false variants. Validation of CRISP on two separate pooled sequencing datasets generated using the Illumina Genome Analyzer demonstrates that it can detect 80-85% of SNPs identified using individual sequencing while achieving a low false discovery rate (3-5%). Comparison with previous methods for pooled SNP detection demonstrates the significantly lower false positive and false negative rates for CRISP.
Implementation of this method is available at http://polymorphism.scripps.edu/~vbansal/software/CRISP/.
新一代测序技术已经能够对整个人类基因组进行测序。然而,对完整基因组的常规重测序仍然不可行。新一代测序仪的巨大容量可以用于对数百至数千个人的特定基因组区域进行测序。基于测序的关联研究目前受到测序平台提供的低多重性的限制。池化测序是研究大人群中稀有变异的一种具有成本效益的方法。为了利用 DNA 池化的力量,从池化测序数据中准确识别序列变异是很重要的。从池化测序中检测稀有变体与从个体测序中检测变体具有不同的挑战。
我们描述了一种新的统计方法 CRISP(用于从池化测序中识别单核苷酸多态性(SNP)的综合读分析),它可以通过两种方法来识别稀有和常见的变体:(i)使用列联表比较多个池之间等位基因计数的分布,以及(ii)评估由于测序错误而单独观察多个非参考碱基调用的概率。该框架还结合了有关正向和反向链之间的读取分布以及池大小的信息,以过滤掉假变体。在使用 Illumina Genome Analyzer 生成的两个独立的池化测序数据集上对 CRISP 的验证表明,它可以检测到使用个体测序识别出的 80-85%的 SNP,同时实现低的假发现率(3-5%)。与用于池化 SNP 检测的先前方法的比较表明,CRISP 的假阳性和假阴性率明显更低。
此方法的实现可在 http://polymorphism.scripps.edu/~vbansal/software/CRISP/ 上获得。