Hao Yangyang, Zhang Pengyue, Xuei Xiaoling, Nakshatri Harikrishna, Edenberg Howard J, Li Lang, Liu Yunlong
Department of Medical and Molecular Genetics, Indiana University School of Medicine, Indianapolis, IN, 46202, USA.
Center for Computational Biology and Bioinformatics, Indiana University School of Medicine, Indianapolis, IN, 46202, USA.
BMC Genomics. 2016 Aug 22;17 Suppl 7(Suppl 7):514. doi: 10.1186/s12864-016-2905-x.
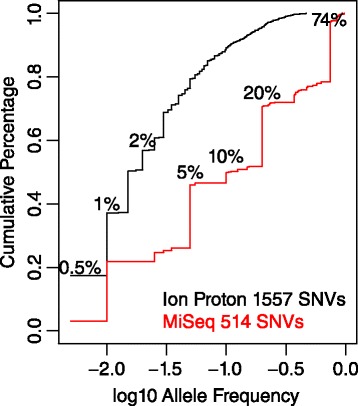
Sensitive detection of low-frequency single nucleotide variants carries great significance in many applications. In cancer genetics research, tumor biopsies are a mixture of normal and tumor cells from various subpopulations due to tumor heterogeneity. Thus the frequencies of somatic variants from a subpopulation tend to be low. Liquid biopsies, which monitor circulating tumor DNA in blood to detect metastatic potential, also face the challenge of detecting low-frequency variants due to the small percentage of the circulating tumor DNA in blood. Moreover, in population genetics research, although pooled sequencing of a large number of individuals is cost-effective, pooling dilutes the signals of variants from any individual. Detection of low frequency variants is difficult and can be cofounded by sequencing artifacts. Existing methods are limited in sensitivity and mainly focus on frequencies around 2 % to 5 %; most fail to consider differential sequencing artifacts.
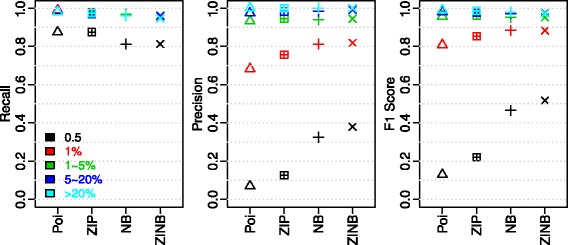
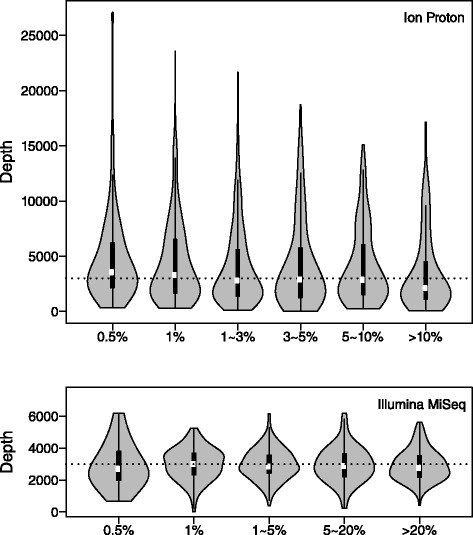
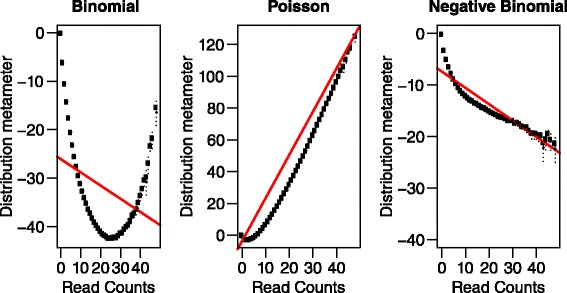
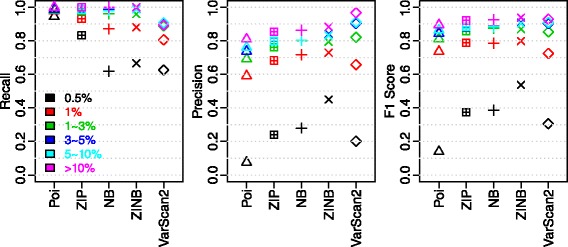
We aimed to push down the frequency detection limit close to the position specific sequencing error rates by modeling the observed erroneous read counts with respect to genomic sequence contexts. 4 distributions suitable for count data modeling (using generalized linear models) were extensively characterized in terms of their goodness-of-fit as well as the performances on real sequencing data benchmarks, which were specifically designed for testing detection of low-frequency variants; two sequencing technologies with significantly different chemistry mechanisms were used to explore systematic errors. We found the zero-inflated negative binomial distribution generalized linear mode is superior to the other models tested, and the advantage is most evident at 0.5 % to 1 % range. This method is also generalizable to different sequencing technologies. Under standard sequencing protocols and depth given in the testing benchmarks, 95.3 % recall and 79.9 % precision for Ion Proton data, 95.6 % recall and 97.0 % precision for Illumina MiSeq data were achieved for SNVs with frequency > = 1 %, while the detection limit is around 0.5 %.
Our method enables sensitive detection of low-frequency single nucleotide variants across different sequencing platforms and will facilitate research and clinical applications such as pooled sequencing, cancer early detection, prognostic assessment, metastatic monitoring, and relapses or acquired resistance identification.
低频单核苷酸变异的灵敏检测在许多应用中具有重要意义。在癌症遗传学研究中,由于肿瘤异质性,肿瘤活检样本是来自不同亚群的正常细胞和肿瘤细胞的混合物。因此,来自亚群的体细胞变异频率往往较低。液体活检通过监测血液中的循环肿瘤DNA来检测转移潜力,但由于循环肿瘤DNA在血液中的比例较小,也面临着检测低频变异的挑战。此外,在群体遗传学研究中,尽管对大量个体进行混合测序具有成本效益,但混合会稀释来自任何个体的变异信号。低频变异的检测困难,并且可能被测序假象所混淆。现有方法在灵敏度方面存在局限性,主要关注2%至5%左右的频率;大多数方法没有考虑差异测序假象。
我们旨在通过对观察到的错误读取计数相对于基因组序列上下文进行建模,将频率检测限降低到接近位置特异性测序错误率。针对适合计数数据建模(使用广义线性模型)的4种分布,在拟合优度以及针对专门设计用于测试低频变异检测的真实测序数据基准的性能方面进行了广泛表征;使用两种具有显著不同化学机制的测序技术来探索系统误差。我们发现零膨胀负二项分布广义线性模型优于其他测试模型,并且在0.5%至1%的范围内优势最为明显。该方法也可推广到不同的测序技术。在测试基准中给定的标准测序方案和深度下,对于频率≥1%的单核苷酸变异,Ion Proton数据的召回率为95.3%,精度为79.9%;Illumina MiSeq数据的召回率为95.6%,精度为97.0%,而检测限约为0.5%。
我们的方法能够灵敏地检测不同测序平台上的低频单核苷酸变异,并将促进诸如混合测序、癌症早期检测、预后评估、转移监测以及复发或获得性耐药鉴定等研究和临床应用。