1National Engineering Research Center for Vegetables, Beijing 100097, China.
BMC Genomics. 2010 Jun 17;11:384. doi: 10.1186/1471-2164-11-384.
Cucumber, Cucumis sativus L., is an economically and nutritionally important crop of the Cucurbitaceae family and has long served as a primary model system for sex determination studies. Recently, the sequencing of its whole genome has been completed. However, transcriptome information of this species is still scarce, with a total of around 8,000 Expressed Sequence Tag (EST) and mRNA sequences currently available in GenBank. In order to gain more insights into molecular mechanisms of plant sex determination and provide the community a functional genomics resource that will facilitate cucurbit research and breeding, we performed transcriptome sequencing of cucumber flower buds of two near-isogenic lines, WI1983G, a gynoecious plant which bears only pistillate flowers, and WI1983H, a hermaphroditic plant which bears only bisexual flowers.
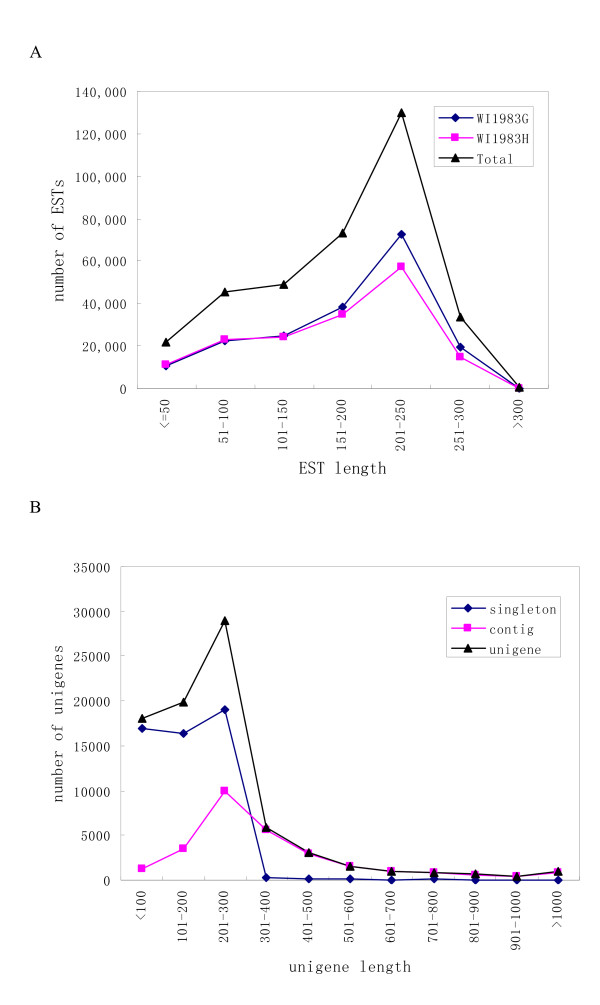
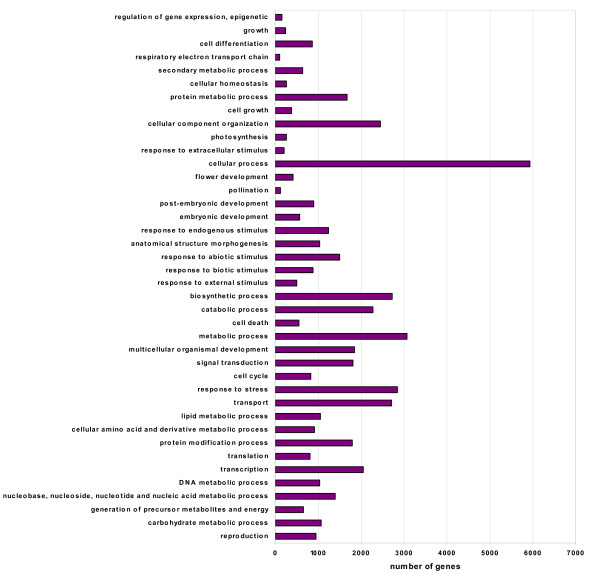
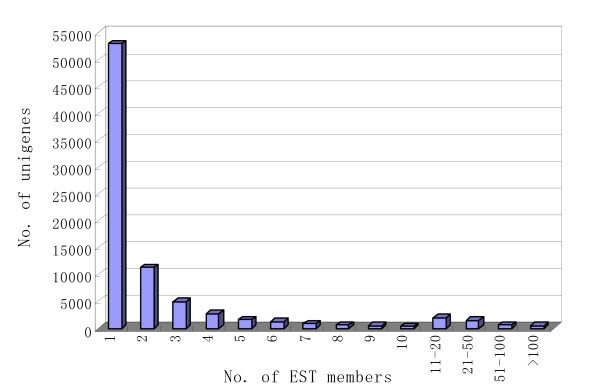
Using Roche-454 massive parallel pyrosequencing technology, we generated a total of 353,941 high quality EST sequences with an average length of 175bp, among which 188,255 were from gynoecious flowers and 165,686 from hermaphroditic flowers. These EST sequences, together with approximately 5,600 high quality cucumber EST and mRNA sequences available in GenBank, were clustered and assembled into 81,401 unigenes, of which 28,452 were contigs and 52,949 were singletons. The unigenes and ESTs were further mapped to the cucumber genome and more than 500 alternative splicing events were identified in 443 cucumber genes. The unigenes were further functionally annotated by comparing their sequences to different protein and functional domain databases and assigned with Gene Ontology (GO) terms. A biochemical pathway database containing 343 predicted pathways was also created based on the annotations of the unigenes. Digital expression analysis identified approximately 200 differentially expressed genes between flowers of WI1983G and WI1983H and provided novel insights into molecular mechanisms of plant sex determination process. Furthermore, a set of SSR motifs and high confidence SNPs between WI1983G and WI1983H were identified from the ESTs, which provided the material basis for future genetic linkage and QTL analysis.
A large set of EST sequences were generated from cucumber flower buds of two different sex types. Differentially expressed genes between these two different sex-type flowers, as well as putative SSR and SNP markers, were identified. These EST sequences provide valuable information to further understand molecular mechanisms of plant sex determination process and forms a rich resource for future functional genomics analysis, marker development and cucumber breeding.
黄瓜(Cucumis sativus L.)是葫芦科中经济和营养上都很重要的作物,长期以来一直是性别决定研究的主要模式系统。最近,其整个基因组的测序已经完成。然而,该物种的转录组信息仍然很少,目前在 GenBank 中总共只有约 8000 个表达序列标签(EST)和 mRNA 序列。为了更深入地了解植物性别决定的分子机制,并为葫芦科研究和育种提供一个功能基因组学资源,我们对两个近等基因系黄瓜花蕾的转录组进行了测序,WI1983G 是一个只产生雌花的雌性植物,WI1983H 是一个只产生两性花的雌雄同体植物。
使用 Roche-454 大规模平行焦磷酸测序技术,我们共生成了 353941 条高质量 EST 序列,平均长度为 175bp,其中 188255 条来自雌性花,165686 条来自两性花。这些 EST 序列,加上 GenBank 中大约 5600 条高质量的黄瓜 EST 和 mRNA 序列,被聚类并组装成 81401 个单基因,其中 28452 个是连续基因,52949 个是单基因。这些单基因和 EST 进一步映射到黄瓜基因组上,在 443 个黄瓜基因中鉴定出 500 多个选择性剪接事件。通过将它们的序列与不同的蛋白质和功能域数据库进行比较,对单基因进行了进一步的功能注释,并赋予了基因本体论(GO)术语。还根据注释创建了一个包含 343 个预测途径的生化途径数据库。数字表达分析在 WI1983G 和 WI1983H 的花之间鉴定出约 200 个差异表达基因,为植物性别决定过程的分子机制提供了新的见解。此外,还从 EST 中鉴定出 WI1983G 和 WI1983H 之间的一组 SSR 基序和高置信度 SNP,为未来的遗传连锁和 QTL 分析提供了物质基础。
从两种不同性别类型的黄瓜花蕾中生成了大量的 EST 序列。鉴定了这两种不同性别类型的花之间差异表达的基因,以及潜在的 SSR 和 SNP 标记。这些 EST 序列为进一步了解植物性别决定过程的分子机制提供了有价值的信息,并为未来的功能基因组学分析、标记开发和黄瓜育种提供了丰富的资源。