Department of Computer Science, Xavier University of Louisiana, 1 Drexel Drive, New Orleans, LA 70125, USA.
BMC Bioinformatics. 2010 Jun 22;11:338. doi: 10.1186/1471-2105-11-338.
Comparative analysis of gene expression profiling of multiple biological categories, such as different species of organisms or different kinds of tissue, promises to enhance the fundamental understanding of the universality as well as the specialization of mechanisms and related biological themes. Grouping genes with a similar expression pattern or exhibiting co-expression together is a starting point in understanding and analyzing gene expression data. In recent literature, gene module level analysis is advocated in order to understand biological network design and system behaviors in disease and life processes; however, practical difficulties often lie in the implementation of existing methods.
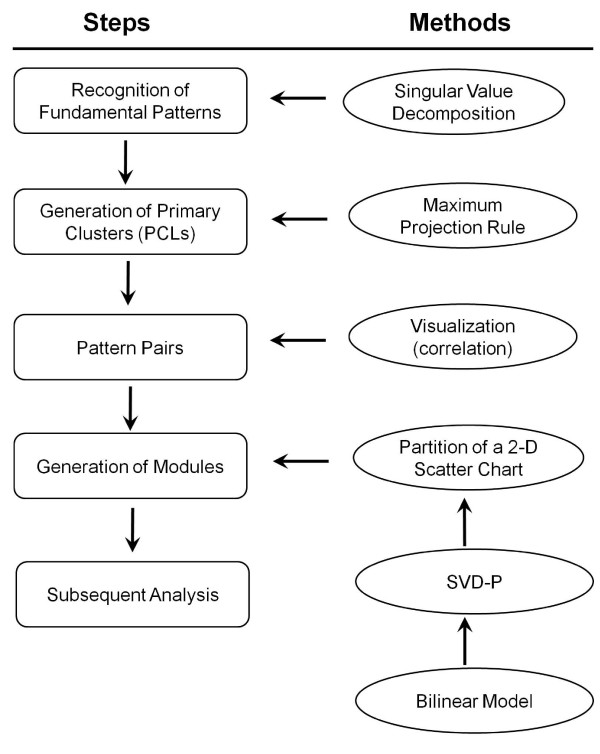
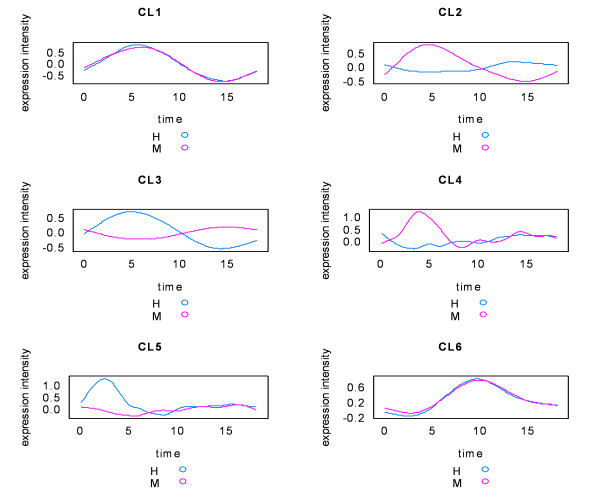
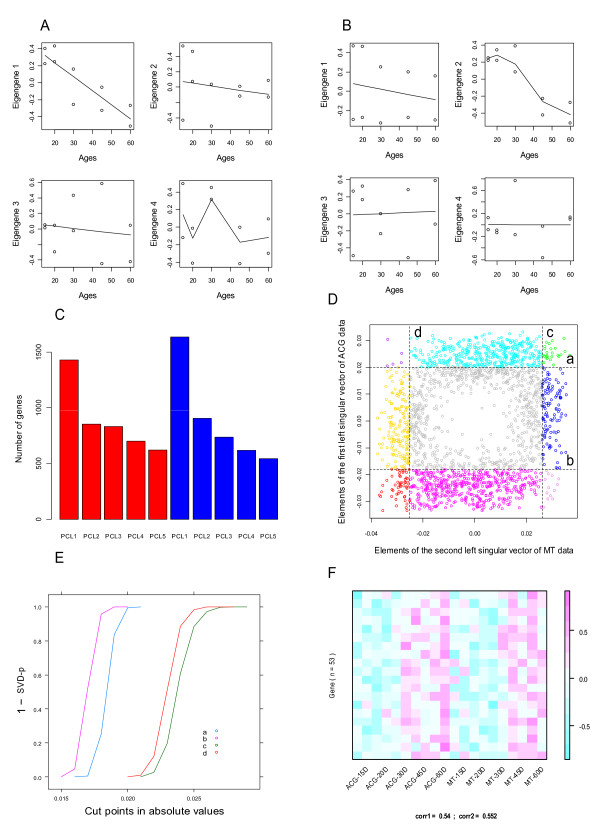
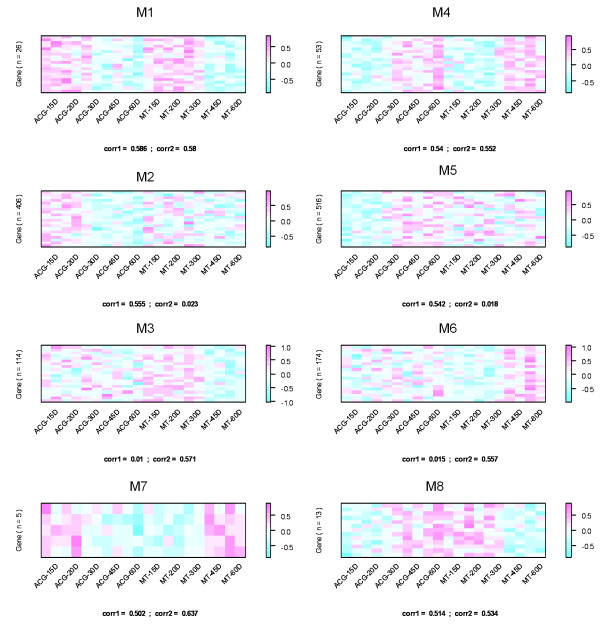
Using the singular value decomposition (SVD) technique, we developed a new computational tool, named svdPPCS (SVD-based Pattern Pairing and Chart Splitting), to identify conserved and divergent co-expression modules of two sets of microarray experiments. In the proposed methods, gene modules are identified by splitting the two-way chart coordinated with a pair of left singular vectors factorized from the gene expression matrices of the two biological categories. Importantly, the cutoffs are determined by a data-driven algorithm using the well-defined statistic, SVD-p. The implementation was illustrated on two time series microarray data sets generated from the samples of accessory gland (ACG) and malpighian tubule (MT) tissues of the line W118 of M. drosophila. Two conserved modules and six divergent modules, each of which has a unique characteristic profile across tissue kinds and aging processes, were identified. The number of genes contained in these models ranged from five to a few hundred. Three to over a hundred GO terms were over-represented in individual modules with FDR < 0.1. One divergent module suggested the tissue-specific relationship between the expressions of mitochondrion-related genes and the aging process. This finding, together with others, may be of biological significance. The validity of the proposed SVD-based method was further verified by a simulation study, as well as the comparisons with regression analysis and cubic spline regression analysis plus PAM based clustering.
svdPPCS is a novel computational tool for the comparative analysis of transcriptional profiling. It especially fits the comparison of time series data of related organisms or different tissues of the same organism under equivalent or similar experimental conditions. The general scheme can be directly extended to the comparisons of multiple data sets. It also can be applied to the integration of data sets from different platforms and of different sources.
对多个生物学类别(例如不同生物体或不同组织)的基因表达谱进行比较分析,有望增强对机制和相关生物学主题普遍性和特殊性的基本理解。将具有相似表达模式或共同表达的基因分组在一起,是理解和分析基因表达数据的起点。在最近的文献中,提倡进行基因模块水平分析,以了解疾病和生命过程中的生物网络设计和系统行为;然而,实际困难往往在于现有方法的实施。
我们使用奇异值分解(SVD)技术开发了一种新的计算工具,名为 svdPPCS(基于 SVD 的模式对配和图表分割),用于识别两组微阵列实验中保守和发散的共表达模块。在提出的方法中,通过分割与从两个生物类别基因表达矩阵的左奇异向量因子化得到的一对左奇异向量协调的双向图表来识别基因模块。重要的是,使用定义明确的统计量 SVD-p 的数据驱动算法确定了截止值。该实施例在来自 M. drosophila 的 W118 品系的附腺(ACG)和马氏管(MT)组织样本生成的两个时间序列微阵列数据集上进行了说明。鉴定了两个保守模块和六个发散模块,每个模块在组织种类和衰老过程中都具有独特的特征谱。这些模型中包含的基因数量从五个到几百个不等。在单个模块中,超过 3 个到 100 多个 GO 术语的过度表达,FDR<0.1。一个发散模块表明了与衰老过程相关的线粒体相关基因表达之间的组织特异性关系。这一发现与其他发现一起,可能具有生物学意义。通过模拟研究以及与回归分析和三次样条回归分析加 PAM 聚类的比较,进一步验证了所提出的基于 SVD 的方法的有效性。
svdPPCS 是一种用于转录谱比较分析的新型计算工具。它特别适合于在相同实验条件下比较相关生物体的时间序列数据或同一生物体的不同组织。通用方案可以直接扩展到多个数据集的比较。它还可以应用于来自不同平台和不同来源的数据集的整合。