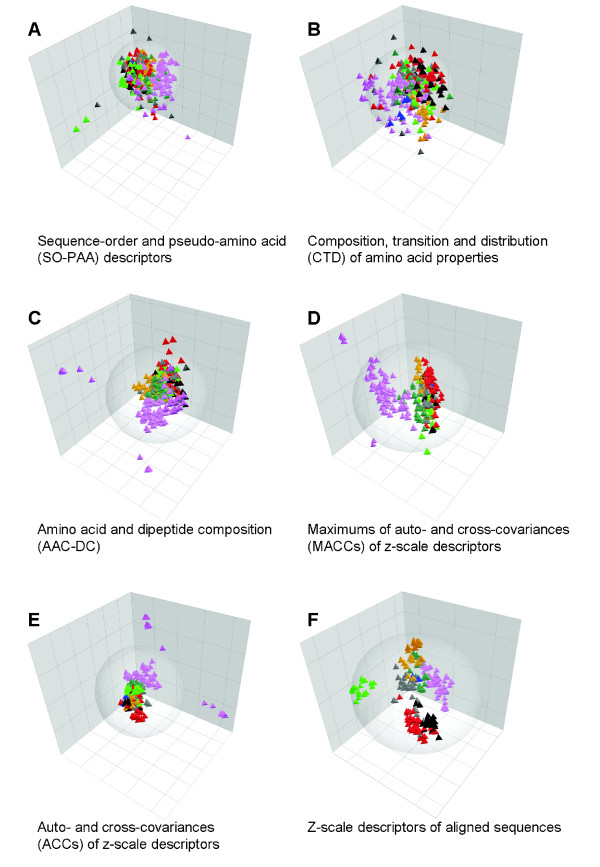
Department of Pharmaceutical Pharmacology, Uppsala University, Sweden.
BMC Bioinformatics. 2010 Jun 22;11:339. doi: 10.1186/1471-2105-11-339.
Protein kinases play crucial roles in cell growth, differentiation, and apoptosis. Abnormal function of protein kinases can lead to many serious diseases, such as cancer. Kinase inhibitors have potential for treatment of these diseases. However, current inhibitors interact with a broad variety of kinases and interfere with multiple vital cellular processes, which causes toxic effects. Bioinformatics approaches that can predict inhibitor-kinase interactions from the chemical properties of the inhibitors and the kinase macromolecules might aid in design of more selective therapeutic agents, that show better efficacy and lower toxicity.
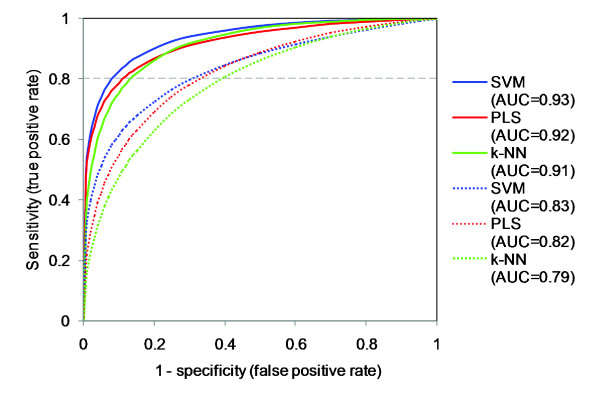
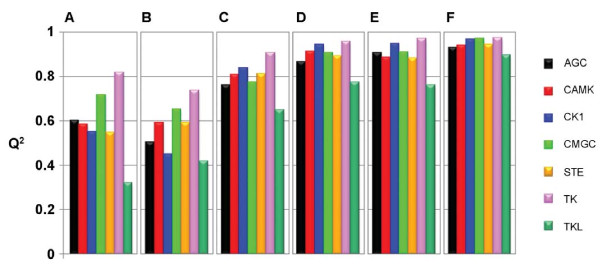
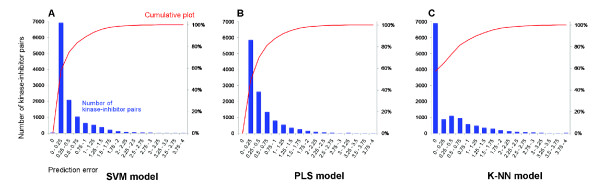
We applied proteochemometric modelling to correlate the properties of 317 wild-type and mutated kinases and 38 inhibitors (12,046 inhibitor-kinase combinations) to the respective combination's interaction dissociation constant (Kd). We compared six approaches for description of protein kinases and several linear and non-linear correlation methods. The best performing models encoded kinase sequences with amino acid physico-chemical z-scale descriptors and used support vector machines or partial least- squares projections to latent structures for the correlations. Modelling performance was estimated by double cross-validation. The best models showed high predictive ability; the squared correlation coefficient for new kinase-inhibitor pairs ranging P2 = 0.67-0.73; for new kinases it ranged P2kin = 0.65-0.70. Models could also separate interacting from non-interacting inhibitor-kinase pairs with high sensitivity and specificity; the areas under the ROC curves ranging AUC = 0.92-0.93. We also investigated the relationship between the number of protein kinases in the dataset and the modelling results. Using only 10% of all data still a valid model was obtained with P2 = 0.47, P2kin = 0.42 and AUC = 0.83.
Our results strongly support the applicability of proteochemometrics for kinome-wide interaction modelling. Proteochemometrics might be used to speed-up identification and optimization of protein kinase targeted and multi-targeted inhibitors.
蛋白激酶在细胞生长、分化和凋亡中起着至关重要的作用。蛋白激酶功能异常可导致许多严重疾病,如癌症。激酶抑制剂具有治疗这些疾病的潜力。然而,目前的抑制剂与多种激酶相互作用,并干扰多种重要的细胞过程,从而导致毒性作用。能够从抑制剂的化学性质和激酶大分子预测抑制剂-激酶相互作用的生物信息学方法,可能有助于设计更具选择性的治疗剂,从而提高疗效和降低毒性。
我们应用蛋白质化学计量模型,将 317 种野生型和突变型激酶和 38 种抑制剂(12046 种抑制剂-激酶组合)的特性与各自组合的相互作用解离常数(Kd)相关联。我们比较了六种描述蛋白激酶的方法和几种线性和非线性相关方法。表现最好的模型编码了激酶序列的氨基酸物理化学 z 标度描述符,并使用支持向量机或偏最小二乘投影到潜在结构进行相关。通过双交叉验证估计模型性能。最好的模型表现出较高的预测能力;新的激酶-抑制剂对的平方相关系数 P2 为 0.67-0.73;新激酶的 P2kin 为 0.65-0.70。模型还可以以较高的灵敏度和特异性将相互作用和非相互作用的抑制剂-激酶对分开;ROC 曲线下的面积 AUC 为 0.92-0.93。我们还研究了数据集内蛋白激酶数量与建模结果之间的关系。仅使用所有数据的 10%,仍然可以获得有效的模型,P2 = 0.47,P2kin = 0.42,AUC = 0.83。
我们的结果强烈支持蛋白质化学计量学在整个激酶组相互作用建模中的适用性。蛋白质化学计量学可用于加速鉴定和优化针对蛋白激酶的和多靶点抑制剂。