Global Pharmaceutical Research and Development, Abbott Laboratories, 100 Abbott Park Road, Building AP-10, Dep. R4CD, Abbott Park, IL 60064, USA.
BMC Med Genomics. 2010 Jun 22;3:23. doi: 10.1186/1755-8794-3-23.
Cancer is a heterogeneous disease caused by genomic aberrations and characterized by significant variability in clinical outcomes and response to therapies. Several subtypes of common cancers have been identified based on alterations of individual cancer genes, such as HER2, EGFR, and others. However, cancer is a complex disease driven by the interaction of multiple genes, so the copy number status of individual genes is not sufficient to define cancer subtypes and predict responses to treatments. A classification based on genome-wide copy number patterns would be better suited for this purpose.
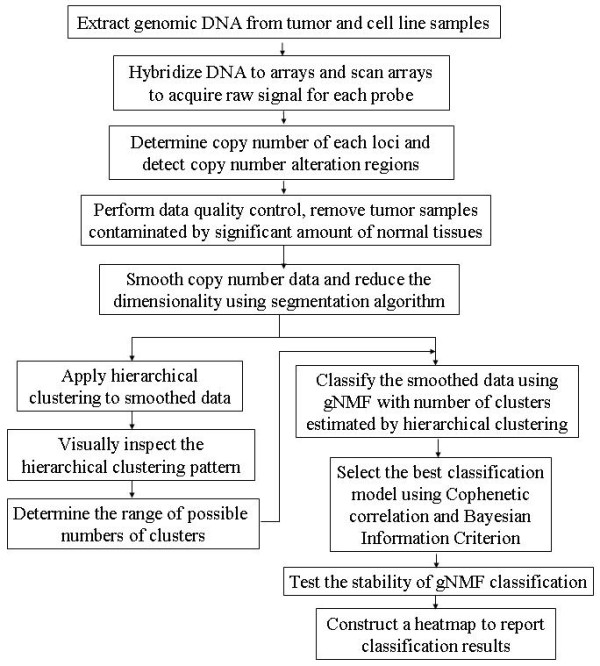
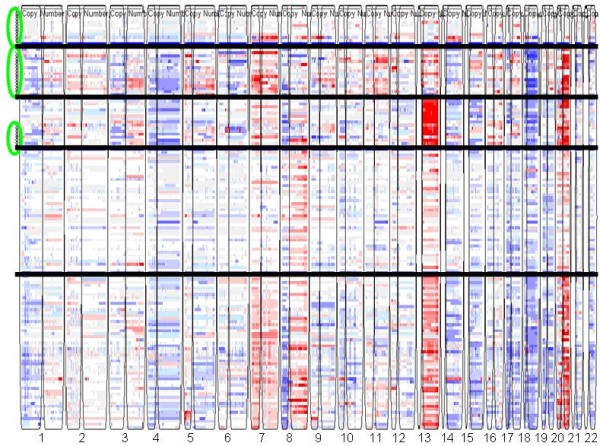
To develop a more comprehensive cancer taxonomy based on genome-wide patterns of copy number abnormalities, we designed an unsupervised classification algorithm that identifies genomic subgroups of tumors. This algorithm is based on a modified genomic Non-negative Matrix Factorization (gNMF) algorithm and includes several additional components, namely a pilot hierarchical clustering procedure to determine the number of clusters, a multiple random initiation scheme, a new stop criterion for the core gNMF, as well as a 10-fold cross-validation stability test for quality assessment.
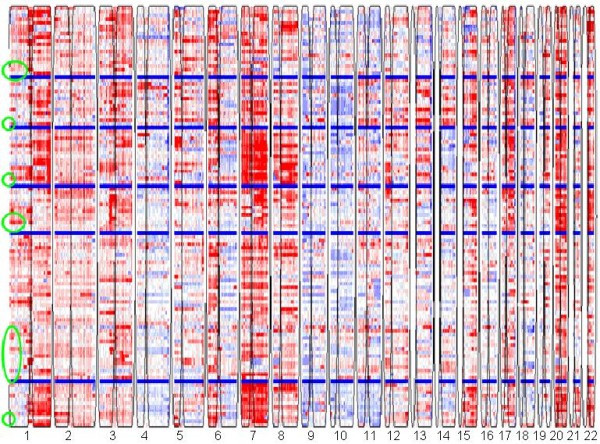
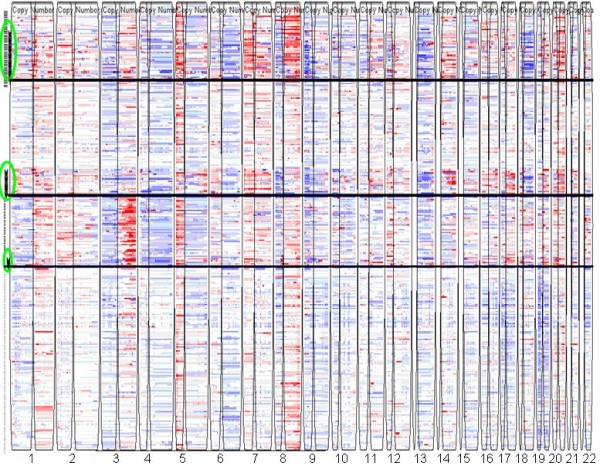
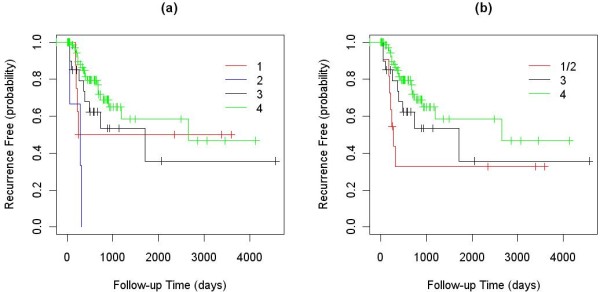
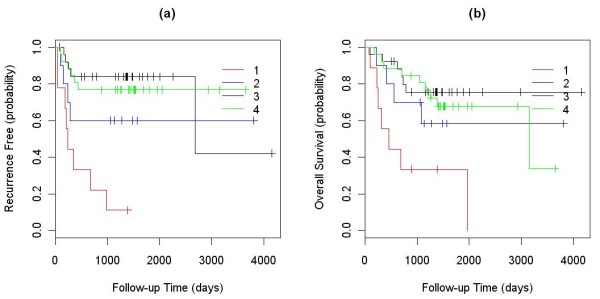
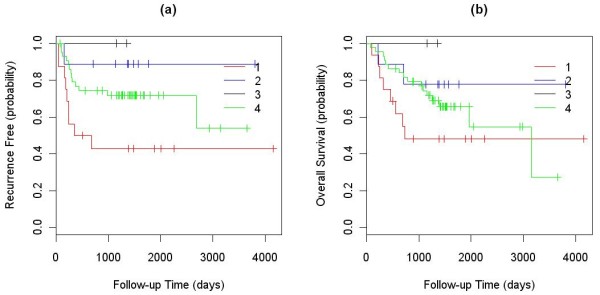
We applied our algorithm to identify genomic subgroups of three major cancer types: non-small cell lung carcinoma (NSCLC), colorectal cancer (CRC), and malignant melanoma. High-density SNP array datasets for patient tumors and established cell lines were used to define genomic subclasses of the diseases and identify cell lines representative of each genomic subtype. The algorithm was compared with several traditional clustering methods and showed improved performance. To validate our genomic taxonomy of NSCLC, we correlated the genomic classification with disease outcomes. Overall survival time and time to recurrence were shown to differ significantly between the genomic subtypes.
We developed an algorithm for cancer classification based on genome-wide patterns of copy number aberrations and demonstrated its superiority to existing clustering methods. The algorithm was applied to define genomic subgroups of three cancer types and identify cell lines representative of these subgroups. Our data enabled the assembly of representative cell line panels for testing drug candidates.
癌症是一种由基因组异常引起的异质性疾病,其临床结果和对治疗的反应存在显著的可变性。已经根据个别癌症基因的改变,如 HER2、EGFR 等,确定了几种常见癌症的亚型。然而,癌症是一种由多个基因相互作用驱动的复杂疾病,因此个别基因的拷贝数状态不足以定义癌症亚型并预测治疗反应。基于全基因组拷贝数模式的分类可能更适合这一目的。
为了基于全基因组拷贝数异常模式开发更全面的癌症分类法,我们设计了一种无监督分类算法,用于识别肿瘤的基因组亚群。该算法基于改进的基因组非负矩阵分解(gNMF)算法,并包含几个附加组件,即用于确定聚类数的试点分层聚类程序、多个随机起始方案、核心 gNMF 的新停止标准,以及用于质量评估的 10 倍交叉验证稳定性测试。
我们应用我们的算法来识别三种主要癌症类型(非小细胞肺癌(NSCLC)、结直肠癌(CRC)和恶性黑色素瘤)的基因组亚群。使用患者肿瘤和已建立的细胞系的高密度 SNP 阵列数据集来定义疾病的基因组亚类,并识别代表每个基因组亚型的细胞系。该算法与几种传统聚类方法进行了比较,显示出了改进的性能。为了验证 NSCLC 的基因组分类法,我们将基因组分类与疾病结果相关联。全生存时间和复发时间在基因组亚型之间存在显著差异。
我们开发了一种基于全基因组拷贝数异常模式的癌症分类算法,并证明了其优于现有聚类方法的性能。该算法应用于定义三种癌症类型的基因组亚群,并识别代表这些亚群的细胞系。我们的数据使代表性细胞系面板能够组装用于测试候选药物。