Mendel Laboratory, Casa Sollievo della Sofferenza Hospital, IRCCS, San Giovanni Rotondo, Italy.
Orphanet J Rare Dis. 2010 Jul 8;5:20. doi: 10.1186/1750-1172-5-20.
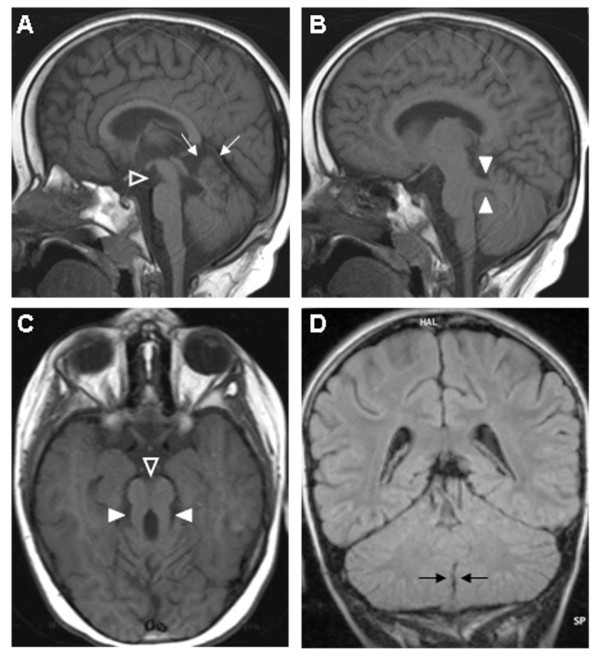
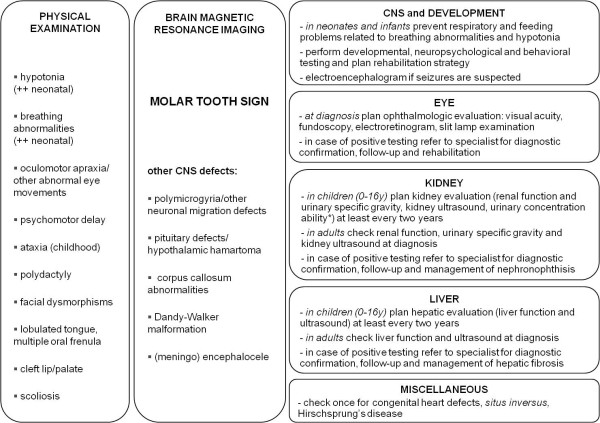
Joubert syndrome (JS) and related disorders (JSRD) are a group of developmental delay/multiple congenital anomalies syndromes in which the obligatory hallmark is the molar tooth sign (MTS), a complex midbrain-hindbrain malformation visible on brain imaging, first recognized in JS. Estimates of the incidence of JSRD range between 1/80,000 and 1/100,000 live births, although these figures may represent an underestimate. The neurological features of JSRD include hypotonia, ataxia, developmental delay, intellectual disability, abnormal eye movements, and neonatal breathing dysregulation. These may be associated with multiorgan involvement, mainly retinal dystrophy, nephronophthisis, hepatic fibrosis and polydactyly, with both inter- and intra-familial variability. JSRD are classified in six phenotypic subgroups: Pure JS; JS with ocular defect; JS with renal defect; JS with oculorenal defects; JS with hepatic defect; JS with orofaciodigital defects. With the exception of rare X-linked recessive cases, JSRD follow autosomal recessive inheritance and are genetically heterogeneous. Ten causative genes have been identified to date, all encoding for proteins of the primary cilium or the centrosome, making JSRD part of an expanding group of diseases called "ciliopathies". Mutational analysis of causative genes is available in few laboratories worldwide on a diagnostic or research basis. Differential diagnosis must consider in particular the other ciliopathies (such as nephronophthisis and Senior-Loken syndrome), distinct cerebellar and brainstem congenital defects and disorders with cerebro-oculo-renal manifestations. Recurrence risk is 25% in most families, although X-linked inheritance should also be considered. The identification of the molecular defect in couples at risk allows early prenatal genetic testing, whereas fetal brain neuroimaging may remain uninformative until the end of the second trimester of pregnancy. Detection of the MTS should be followed by a diagnostic protocol to assess multiorgan involvement. Optimal management requires a multidisciplinary approach, with particular attention to respiratory and feeding problems in neonates and infants. Cognitive and behavioral assessments are also recommended to provide young patients with adequate neuropsychological support and rehabilitation. After the first months of life, global prognosis varies considerably among JSRD subgroups, depending on the extent and severity of organ involvement.
杰伯综合征(JS)和相关疾病(JSRD)是一组发育迟缓/多发先天畸形综合征,其强制性特征是磨牙齿征(MTS),这是一种在脑成像上可见的复杂中脑-后脑畸形,最初在 JS 中被识别。JSRD 的发病率估计在每 80,000 至 100,000 活产儿中 1 例之间,尽管这些数字可能代表低估。JSRD 的神经学特征包括低张力、共济失调、发育迟缓、智力障碍、眼球运动异常和新生儿呼吸失调。这些可能与多器官受累有关,主要是视网膜营养不良、肾单位肾病变、肝纤维化和多指(趾)畸形,具有内外家族变异性。JSRD 分为六个表型亚组:纯 JS;伴有眼部缺陷的 JS;伴有肾脏缺陷的 JS;伴有眼肾缺陷的 JS;伴有肝脏缺陷的 JS;伴有口面指(趾)畸形的 JS。除了罕见的 X 连锁隐性病例外,JSRD 遵循常染色体隐性遗传,遗传异质性。迄今为止,已经确定了 10 个致病基因,这些基因均编码初级纤毛或中心体的蛋白,使 JSRD 成为称为“纤毛病”的不断扩大的疾病组的一部分。目前,全球只有少数实验室可在诊断或研究基础上进行致病基因的突变分析。鉴别诊断必须特别考虑其他纤毛病(如肾单位肾病变和 Senior-Loken 综合征)、不同的小脑和脑干先天性缺陷以及具有脑-眼-肾表现的疾病。大多数家庭的复发风险为 25%,尽管也应考虑 X 连锁遗传。在有风险的夫妇中确定分子缺陷可进行早期产前基因检测,而胎儿脑神经影像学可能直到妊娠中期末仍无信息。MTS 的检测应遵循评估多器官受累的诊断方案。最佳管理需要多学科方法,特别注意新生儿和婴儿的呼吸和喂养问题。还建议进行认知和行为评估,为年轻患者提供足够的神经心理学支持和康复。在生命的头几个月后,JSRD 亚组之间的总体预后差异很大,这取决于器官受累的程度和严重程度。