Department of Bioinformatics and Genomics, Center for Bioinformatics Research, the University of North Carolina at Charlotte, Charlotte, NC 28223, USA.
BMC Bioinformatics. 2010 Jul 23;11:397. doi: 10.1186/1471-2105-11-397.
Our current understanding of transcription factor binding sites (TFBSs) in sequenced prokaryotic genomes is very limited due to the lack of an accurate and efficient computational method for the prediction of TFBSs at a genome scale. In an attempt to change this situation, we have recently developed a comparative genomics based algorithm called GLECLUBS for de novo genome-wide prediction of TFBSs in a target genome. Although GLECLUBS has achieved rather high prediction accuracy of TFBSs in a target genome, it is still not efficient enough to be applied to all the sequenced prokaryotic genomes.
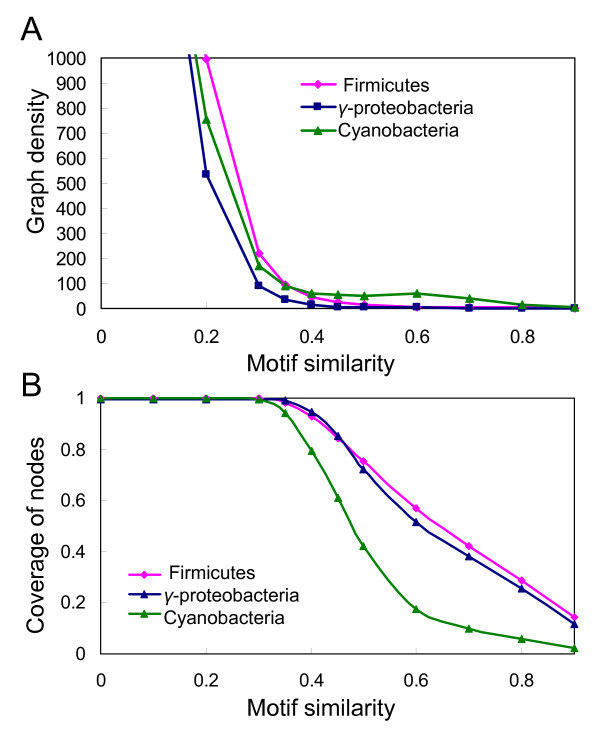
Here, we designed a new algorithm based on GLECLUBS called extended GLECLUBS (eGLECLUBS) for simultaneous prediction of TFBSs in a group of related prokaryotic genomes. When tested on a group of gamma-proteobacterial genomes including E. coli K12, a group of firmicutes genomes including B. subtilis and a group of cyanobacterial genomes using the same parameter settings, eGLECLUBS predicts more than 82% of known TFBSs in extracted inter-operonic sequences in both E. coli K12 and B. subtilis. Because each genome in a group is equally treated, it is highly likely that similar prediction accuracy has been achieved for each genome in the group.
We have developed a new algorithm for genome-wide de novo prediction of TFBSs in a group of related prokaryotic genomes. The algorithm has achieved the same level of accuracy and robustness as its predecessor GLECLUBS, but can work on dozens of genomes at the same time.
由于缺乏一种准确有效的计算方法来预测基因组范围内的转录因子结合位点(TFBS),我们目前对测序原核基因组中的 TFBS 的理解非常有限。为了改变这种情况,我们最近开发了一种基于比较基因组学的算法,称为 GLECLUBS,用于在目标基因组中进行 TFBS 的从头全基因组预测。尽管 GLECLUBS 在目标基因组中实现了相当高的 TFBS 预测准确性,但它的效率仍然不够高,无法应用于所有测序的原核基因组。
在这里,我们设计了一种基于 GLECLUBS 的新算法,称为扩展 GLECLUBS(eGLECLUBS),用于同时预测一组相关原核基因组中的 TFBS。当使用相同的参数设置在包括 E. coli K12 的一组γ-变形杆菌基因组、包括 B. subtilis 的一组Firmicutes 基因组和一组蓝藻基因组上进行测试时,eGLECLUBS 在提取的 E. coli K12 和 B. subtilis 中的种间序列中预测了超过 82%的已知 TFBS。由于组中的每个基因组都被平等对待,因此很可能在组中的每个基因组中都实现了类似的预测准确性。
我们已经开发了一种新的算法,用于在一组相关的原核基因组中进行全基因组从头预测 TFBS。该算法与其前身 GLECLUBS 具有相同的准确性和稳健性,但可以同时处理几十个基因组。