Zhang Shaoqiang, Zhou Xiguo, Du Chuanbin, Su Zhengchang
BMC Syst Biol. 2013;7 Suppl 2(Suppl 2):S14. doi: 10.1186/1752-0509-7-S2-S14. Epub 2013 Dec 17.
Discovering transcription factor binding sites (TFBS) is one of primary challenges to decipher complex gene regulatory networks encrypted in a genome. A set of short DNA sequences identified by a transcription factor (TF) is known as a motif, which can be expressed accurately in matrix form such as a position-specific scoring matrix (PSSM) and a position frequency matrix. Very frequently, we need to query a motif in a database of motifs by seeking its similar motifs, merge similar TFBS motifs possibly identified by the same TF, separate irrelevant motifs, or filter out spurious motifs. Therefore, a novel metric is required to seize slight differences between irrelevant motifs and highlight the similarity between motifs of the same group in all these applications. While there are already several metrics for motif similarity proposed before, their performance is still far from satisfactory for these applications.
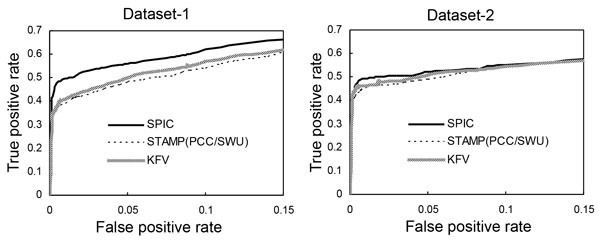
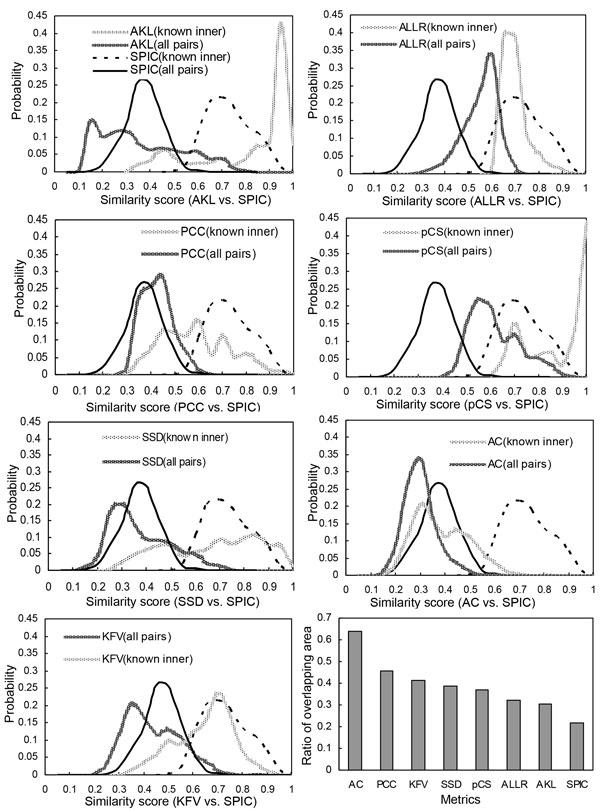
A novel metric has been proposed in this paper with name as SPIC (Similarity with Position Information Contents) for measuring the similarity between a column of a motif and a column of another motif. When defining this similarity score, we consider the likelihood that the column of the first motif's PFM can be produced by the column of the second motif's PSSM, and multiply the likelihood by the information content of the column of the second motif's PSSM, and vise versa. We evaluated the performance of SPIC combined with a local or a global alignment method having a function for affine gap penalty, for computing the similarity between two motifs. We also compared SPIC with seven existing state-of-the-arts metrics for their capability of clustering motifs from the same group and retrieving motifs from a database on three datasets.
When used jointly with the Smith-Waterman local alignment method with an affine gap penalty function (gap open penalty is equal to 1, gap extension penalty is equal to 0.5), SPIC outperforms the seven existing state-of-the-art motif similarity metrics combined with their best alignments for matching motifs in database searches, and clustering the same TF's sub-motifs or distinguishing relevant ones from a miscellaneous group of motifs.
We have developed a novel motif similarity metric that can more accurately match motifs in database searches, and more effectively cluster similar motifs and differentiate irrelevant motifs than do the other seven metrics we are aware of.
发现转录因子结合位点(TFBS)是解读基因组中加密的复杂基因调控网络的主要挑战之一。由转录因子(TF)识别的一组短DNA序列被称为基序,它可以准确地以矩阵形式表示,如位置特异性评分矩阵(PSSM)和位置频率矩阵。我们经常需要通过寻找相似基序在基序数据库中查询一个基序,合并可能由同一TF识别的相似TFBS基序,分离不相关的基序,或过滤掉假基序。因此,需要一种新的度量标准来捕捉不相关基序之间的细微差异,并在所有这些应用中突出同一组基序之间的相似性。虽然之前已经提出了几种基序相似性度量标准,但它们在这些应用中的性能仍然远不能令人满意。
本文提出了一种名为SPIC(带位置信息含量的相似性)的新度量标准,用于测量一个基序的一列与另一个基序的一列之间的相似性。在定义这个相似性分数时,我们考虑第一个基序的PFM的列由第二个基序的PSSM的列产生的可能性,并将该可能性乘以第二个基序的PSSM的列的信息含量,反之亦然。我们评估了结合具有仿射间隙罚分功能的局部或全局比对方法的SPIC在计算两个基序之间相似性方面的性能。我们还将SPIC与七种现有的最先进的度量标准进行了比较,比较它们在三个数据集上对同一组基序进行聚类以及从数据库中检索基序的能力。
当与具有仿射间隙罚分函数(间隙开放罚分等于1,间隙延伸罚分等于0.5)的Smith-Waterman局部比对方法联合使用时,SPIC在数据库搜索中匹配基序、对同一TF的子基序进行聚类或从一组混杂的基序中区分相关基序方面,优于七种现有的最先进的基序相似性度量标准及其最佳比对。
我们开发了一种新的基序相似性度量标准,与我们所知的其他七种度量标准相比,它在数据库搜索中能更准确地匹配基序,更有效地对相似基序进行聚类并区分不相关基序。