Division of Cancer Treatment and Diagnosis, National Cancer Institute, National Institutes of Health, Bethesda, Maryland, USA.
J Transl Med. 2010 Jul 21;8:69. doi: 10.1186/1479-5876-8-69.
MiR arrays distinguish themselves from gene expression arrays by their more limited number of probes, and the shorter and less flexible sequence in probe design. Robust data processing and analysis methods tailored to the unique characteristics of miR arrays are greatly needed. Assumptions underlying commonly used normalization methods for gene expression microarrays containing tens of thousands or more probes may not hold for miR microarrays. Findings from previous studies have sometimes been inconclusive or contradictory. Further studies to determine optimal normalization methods for miR microarrays are needed.
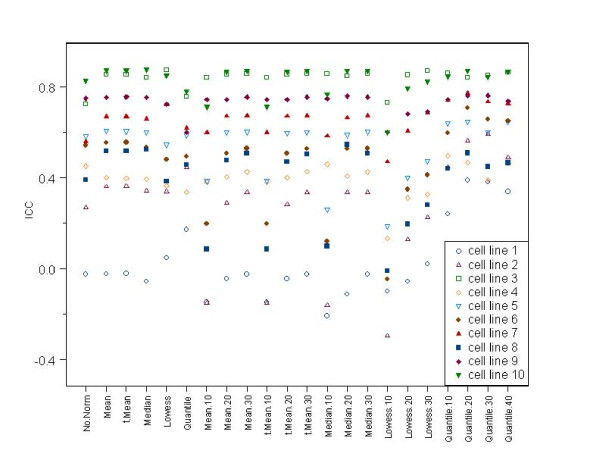
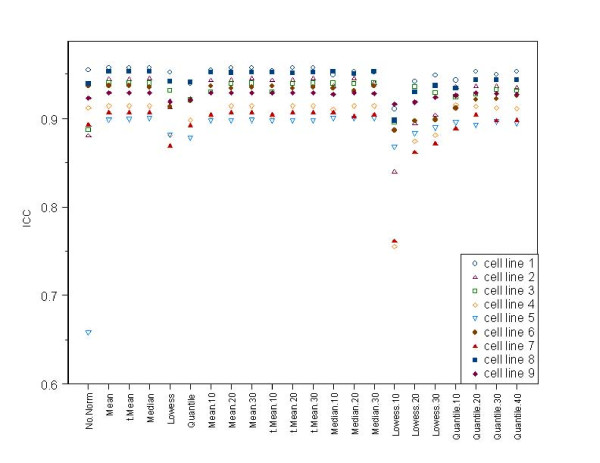
We evaluated many different normalization methods for data generated with a custom-made two channel miR microarray using two data sets that have technical replicates from several different cell lines. The impact of each normalization method was examined on both within miR error variance (between replicate arrays) and between miR variance to determine which normalization methods minimized differences between replicate samples while preserving differences between biologically distinct miRs.
Lowess normalization generally did not perform as well as the other methods, and quantile normalization based on an invariant set showed the best performance in many cases unless restricted to a very small invariant set. Global median and global mean methods performed reasonably well in both data sets and have the advantage of computational simplicity.
Researchers need to consider carefully which assumptions underlying the different normalization methods appear most reasonable for their experimental setting and possibly consider more than one normalization approach to determine the sensitivity of their results to normalization method used.
miRNA 芯片在探针数量、探针设计的序列长度和灵活性方面与基因表达芯片有所不同。因此,非常需要针对 miRNA 芯片独特特点而定制的稳健的数据处理和分析方法。对于包含数万甚至更多探针的基因表达微阵列,常用的标准化方法所基于的假设可能不适用于 miRNA 微阵列。先前的研究结果有时并不明确或相互矛盾。需要进一步研究以确定 miRNA 微阵列的最佳标准化方法。
我们使用两个包含来自多个不同细胞系的技术重复的数据集,评估了一种定制的双通道 miRNA 微阵列生成的数据的多种不同标准化方法。检查了每种标准化方法对 miRNA 内误差方差(重复阵列之间)和 miRNA 之间方差的影响,以确定哪种标准化方法在最小化重复样本之间差异的同时保留了生物学上不同的 miRNAs 之间的差异。
在许多情况下,除非限制在非常小的不变集,否则 Lowess 标准化通常不如其他方法表现出色,而基于不变集的分位数标准化在许多情况下表现最佳。在两个数据集上,全局中位数和全局均值方法的性能都相当不错,并且具有计算简单的优点。
研究人员需要仔细考虑不同标准化方法所基于的假设在其实验设置中最为合理,并且可能需要考虑多种标准化方法来确定其结果对使用的标准化方法的敏感性。