Biological Sciences Division, University of Chicago, Chicago, Illinois, United States of America.
PLoS One. 2010 Jul 29;5(7):e11652. doi: 10.1371/journal.pone.0011652.
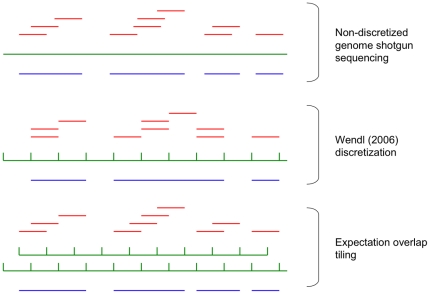
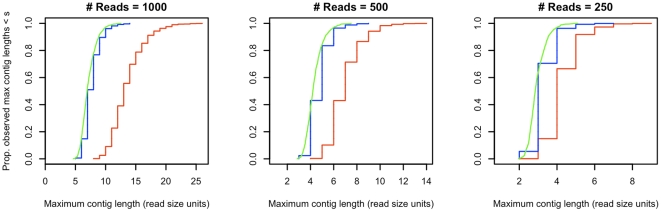
Mathematical aspects of coverage and gaps in genome assembly have received substantial attention by bioinformaticians. Typical problems under consideration suppose that reads can be experimentally obtained from a single genome and that the number of reads will be set to cover a large percentage of that genome at a desired depth. In metagenomics experiments genomes from multiple species are simultaneously analyzed and obtaining large numbers of reads per genome is unlikely. We propose the probability of obtaining at least one contig of a desired minimum size from each novel genome in the pool without restriction based on depth of coverage as a metric for metagenomic experimental design. We derive an approximation to the distribution of maximum contig size for single genome assemblies using relatively few reads. This approximation is verified in simulation studies and applied to a number of different metagenomic experimental design problems, ranging in difficulty from detecting a single novel genome in a pool of known species to detecting each of a random number of novel genomes collectively sized and with abundances corresponding to given distributions in a single pool.
基因组组装的覆盖范围和缺口的数学方面受到了生物信息学家的广泛关注。典型的考虑问题是假设可以从单个基因组中实验获得读取,并且读取的数量将设置为以所需的深度覆盖该基因组的很大一部分。在宏基因组学实验中,同时分析多个物种的基因组,并且不太可能为每个基因组获得大量的读取。我们提出了一种基于覆盖深度的方法,在不限制的情况下,从池中每个新基因组中获得至少一个所需最小大小的连续体的概率作为宏基因组实验设计的度量标准。我们使用相对较少的读取来推导出单基因组组装的最大连续体大小的分布的近似值。该近似值在模拟研究中得到验证,并应用于许多不同的宏基因组实验设计问题,从检测池中的单个新基因组到检测单个池中随机数量的新基因组的集合,这些基因组的大小和丰度对应于给定的分布。