Wendl Michael C, Kota Karthik, Weinstock George M, Mitreva Makedonka
The Genome Institute, Washington University, St. Louis, MO, 63108, USA,
J Math Biol. 2013 Nov;67(5):1141-61. doi: 10.1007/s00285-012-0586-x. Epub 2012 Sep 11.
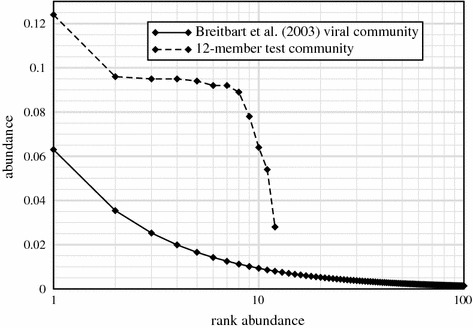
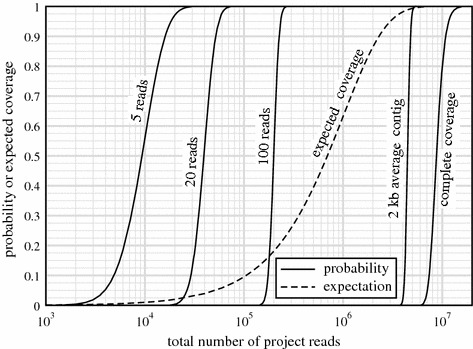
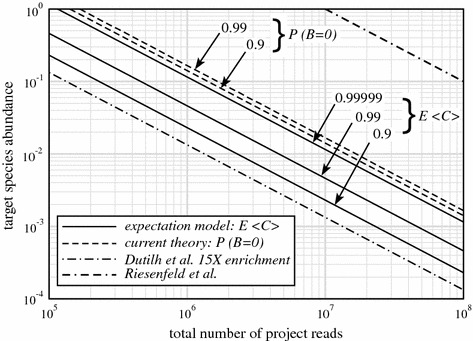
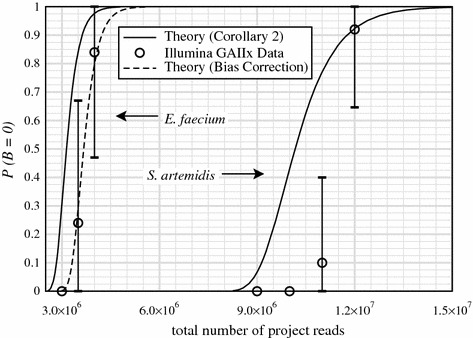
Metagenomic project design has relied variously upon speculation, semi-empirical and ad hoc heuristic models, and elementary extensions of single-sample Lander-Waterman expectation theory, all of which are demonstrably inadequate. Here, we propose an approach based upon a generalization of Stevens' Theorem for randomly covering a domain. We extend this result to account for the presence of multiple species, from which are derived useful probabilities for fully recovering a particular target microbe of interest and for average contig length. These show improved specificities compared to older measures and recommend deeper data generation than the levels chosen by some early studies, supporting the view that poor assemblies were due at least somewhat to insufficient data. We assess predictions empirically by generating roughly 4.5 Gb of sequence from a twelve member bacterial community, comparing coverage for two particular members, Selenomonas artemidis and Enterococcus faecium, which are the least ([Formula: see text]3 %) and most ([Formula: see text]12 %) abundant species, respectively. Agreement is reasonable, with differences likely attributable to coverage biases. We show that, in some cases, bias is simple in the sense that a small reduction in read length to simulate less efficient covering brings data and theory into essentially complete accord. Finally, we describe two applications of the theory. One plots coverage probability over the relevant parameter space, constructing essentially a "metagenomic design map" to enable straightforward analysis and design of future projects. The other gives an overview of the data requirements for various types of sequencing milestones, including a desired number of contact reads and contig length, for detection of a rare viral species.
宏基因组项目设计在不同程度上依赖于推测、半经验和临时启发式模型,以及单样本兰德-沃特曼期望理论的基本扩展,而所有这些方法显然都存在不足。在此,我们提出一种基于史蒂文斯定理推广的方法,用于随机覆盖一个区域。我们将这一结果进行扩展,以考虑多种物种的存在,从中推导出用于完全恢复特定目标微生物以及平均重叠群长度的有用概率。与以往的测量方法相比,这些概率显示出更高的特异性,并建议生成比一些早期研究选择的深度更深的数据,这支持了这样一种观点,即组装效果不佳至少在一定程度上是由于数据不足。我们通过从一个由12种细菌组成的群落中生成约4.5Gb的序列来实证评估预测结果,比较了两种特定成员(分别是丰度最低(约3%)的阿氏月形单胞菌和丰度最高(约12%)的粪肠球菌)的覆盖情况。二者吻合度合理,差异可能归因于覆盖偏差。我们表明,在某些情况下,偏差很简单,即通过小幅缩短读长来模拟效率较低的覆盖,能使数据与理论基本达成一致。最后,我们描述了该理论的两个应用。一个是在相关参数空间上绘制覆盖概率,本质上构建一个“宏基因组设计图”,以便对未来项目进行直接分析和设计。另一个是概述了实现各种测序里程碑(包括检测一种罕见病毒物种所需的接触读段数量和重叠群长度)的数据要求。