Department of Physics, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213, USA.
J Am Chem Soc. 2010 Sep 29;132(38):13129-31. doi: 10.1021/ja105206w.
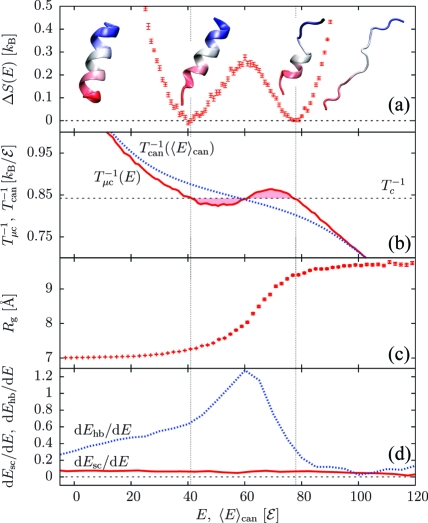
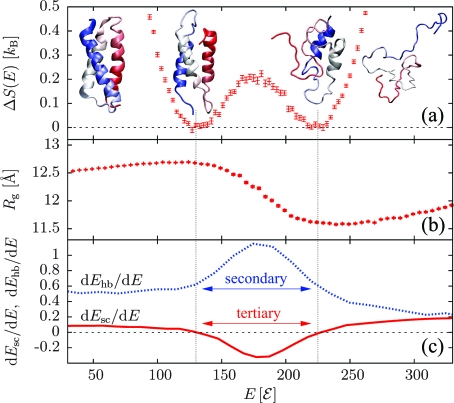
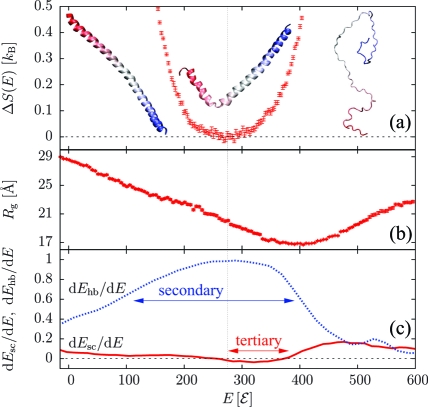
Protein folding cooperativity is defined by the nature of the finite-size thermodynamic transition exhibited upon folding: two-state transitions show a free-energy barrier between the folded and unfolded ensembles, while downhill folding is barrierless. A microcanonical analysis, where the energy is the natural variable, has proved to be better suited than its canonical counterpart to unambiguously characterize the nature of the transition. Replica-exchange molecular dynamics simulations of a high-resolution coarse-grained model allow for the accurate evaluation of the density of states in order to extract precise thermodynamic information and measure its impact on structural features. The method has been applied to three helical peptides: a short helix shows sharp features of a two-state folder, while a longer helix and a three-helix bundle exhibit downhill and two-state transitions, respectively. Extending the results of lattice simulations and theoretical models, we have found that it is the interplay between secondary structure and the loss of non-native tertiary contacts that determines the nature of the transition.
两态转变在折叠和未折叠的集合之间表现出自由能势垒,而顺境折叠则没有势垒。微正则分析,其中能量是自然变量,已被证明比其正则对应物更适合于明确表征转变的性质。高分辨率粗粒模型的 replica-exchange 分子动力学模拟允许准确评估态密度,以提取精确的热力学信息并测量其对结构特征的影响。该方法已应用于三种螺旋肽:短螺旋表现出两态折叠的鲜明特征,而较长的螺旋和三螺旋束分别表现出顺境和两态转变。扩展了晶格模拟和理论模型的结果,我们发现决定转变性质的是二级结构和非天然三级结构丧失之间的相互作用。