Department of Biology, Harvey Mudd College, Claremont, CA, USA.
BMC Bioinformatics. 2010 Sep 15;11:462. doi: 10.1186/1471-2105-11-462.
Models of sequence evolution typically assume that different nucleotide positions evolve independently. This assumption is widely appreciated to be an over-simplification. The best known violations involve biases due to adjacent nucleotides. There have also been suggestions that biases exist at larger scales, however this possibility has not been systematically explored.
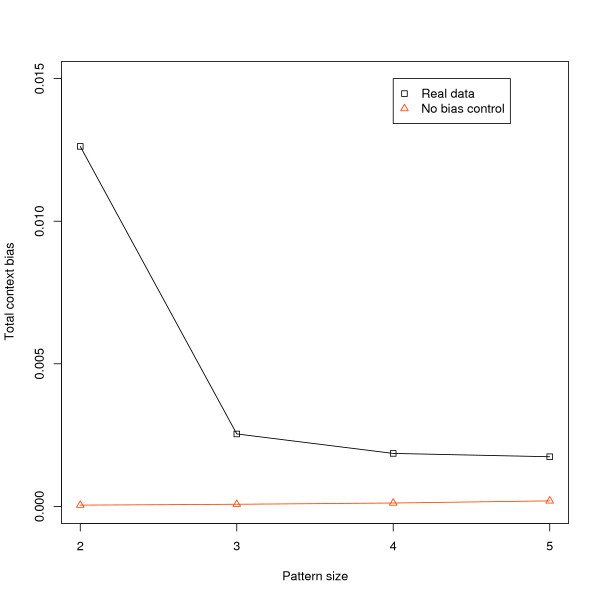
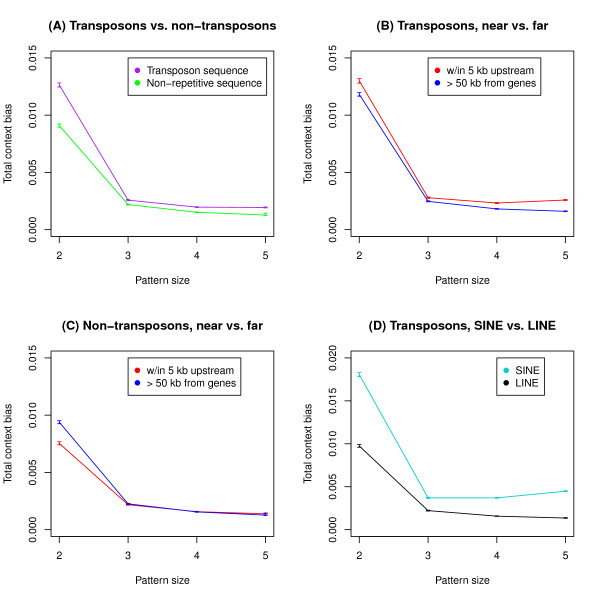
To address this we have developed a method which identifies over- and under-represented substitution patterns and assesses their overall impact on the evolution of genome composition. Our method is designed to account for biases at smaller pattern sizes, removing their effects. We used this method to investigate context bias in the human lineage after the divergence from chimpanzee. We examined bias effects in substitution patterns between 2 and 5 bp long and found significant effects at all sizes. This included some individual three and four base pair patterns with relatively large biases. We also found that bias effects vary across the genome, differing between transposons and non-transposons, between different classes of transposons, and also near and far from genes.
We found that nucleotides beyond the immediately adjacent one are responsible for substantial context effects, and that these biases vary across the genome.
序列进化模型通常假设不同的核苷酸位置是独立进化的。这种假设被广泛认为是过于简化的。最著名的违反情况涉及由于相邻核苷酸引起的偏差。也有人提出在更大的尺度上存在偏差,但这种可能性尚未得到系统的探索。
为了解决这个问题,我们开发了一种方法,可以识别过度和不足的替代模式,并评估它们对基因组组成进化的总体影响。我们的方法旨在考虑较小模式大小的偏差,并消除其影响。我们使用这种方法来研究人类与黑猩猩分化后的人类谱系中的上下文偏差。我们检查了 2 到 5 个碱基长的替代模式中的偏差效应,并在所有大小上都发现了显著的效应。这包括一些具有相对较大偏差的个别三个和四个碱基对模式。我们还发现,偏差效应在整个基因组中有所不同,在转座子和非转座子之间、不同类型的转座子之间以及基因附近和远处都有所不同。
我们发现,紧邻的核苷酸之外的核苷酸负责大量的上下文效应,并且这些偏差在整个基因组中有所不同。