Department of Bioinformatics, Institute of Microbiology and Genetics, Georg-August-University Göttingen, Germany.
BMC Bioinformatics. 2010 Sep 24;11:481. doi: 10.1186/1471-2105-11-481.
Establishing the relationship between an organism's genome sequence and its phenotype is a fundamental challenge that remains largely unsolved. Accurately predicting microbial phenotypes solely based on genomic features will allow us to infer relevant phenotypic characteristics when the availability of a genome sequence precedes experimental characterization, a scenario that is favored by the advent of novel high-throughput and single cell sequencing techniques.
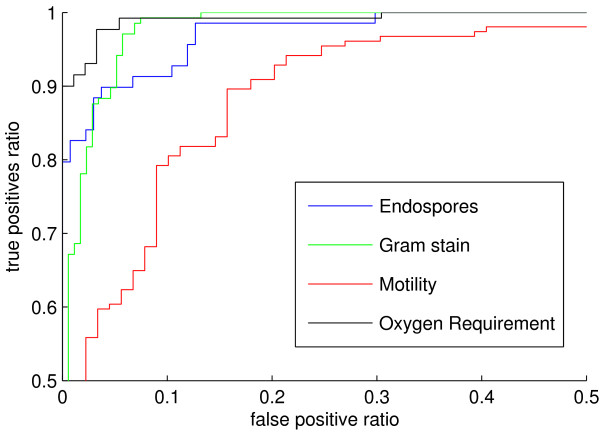
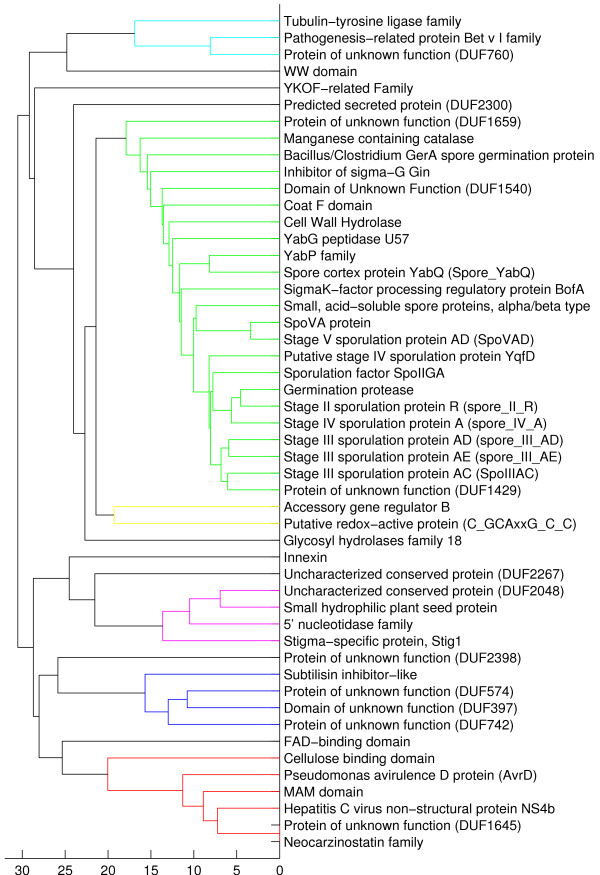
We present a novel approach to predict the phenotype of prokaryotes directly from their protein domain frequencies. Our discriminative machine learning approach provides high prediction accuracy of relevant phenotypes such as motility, oxygen requirement or spore formation. Moreover, the set of discriminative domains provides biological insight into the underlying phenotype-genotype relationship and enables deriving hypotheses on the possible functions of uncharacterized domains.
Fast and accurate prediction of microbial phenotypes based on genomic protein domain content is feasible and has the potential to provide novel biological insights. First results of a systematic check for annotation errors indicate that our approach may also be applied to semi-automatic correction and completion of the existing phenotype annotation.
建立生物体基因组序列与其表型之间的关系是一个基本挑战,目前尚未得到很好的解决。仅基于基因组特征准确预测微生物表型,当可用的基因组序列先于实验特征描述时,我们可以推断出相关的表型特征,这种情况在新型高通量和单细胞测序技术出现后变得有利。
我们提出了一种从蛋白质结构域频率直接预测原核生物表型的新方法。我们的判别机器学习方法提供了对相关表型(如运动性、需氧性或孢子形成)的高预测准确性。此外,这些判别结构域集为潜在的表型-基因型关系提供了生物学见解,并能够推导出关于未表征结构域可能功能的假设。
基于基因组蛋白质结构域含量快速准确地预测微生物表型是可行的,并且有可能提供新的生物学见解。对注释错误进行系统检查的初步结果表明,我们的方法也可应用于半自动校正和完成现有表型注释。