Programa de Estudio y Control de Enfermedades Tropicales-PECET, Universidad de Antioquia, Calle 62 No 52-59, Lab. 632, Medellín, Colombia.
BMC Bioinformatics. 2010 Sep 27;11:484. doi: 10.1186/1471-2105-11-484.
Leishmaniasis is a virulent parasitic infection that causes a worldwide disease burden. Most treatments have toxic side-effects and efficacy has decreased due to the emergence of resistant strains. The outlook is worsened by the absence of promising drug targets for this disease. We have taken a computational approach to the detection of new drug targets, which may become an effective strategy for the discovery of new drugs for this tropical disease.
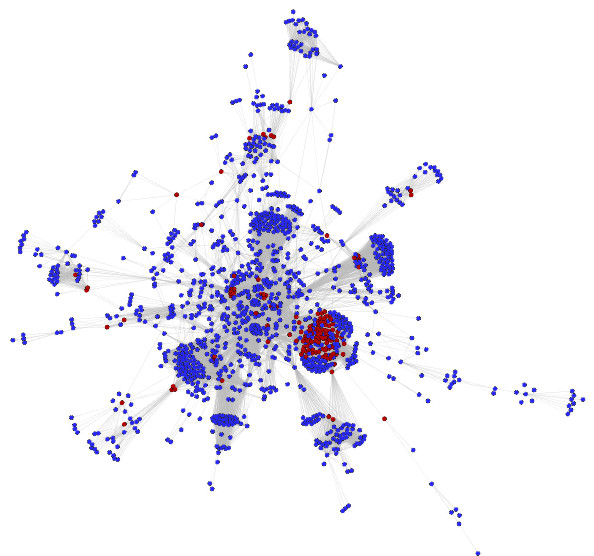
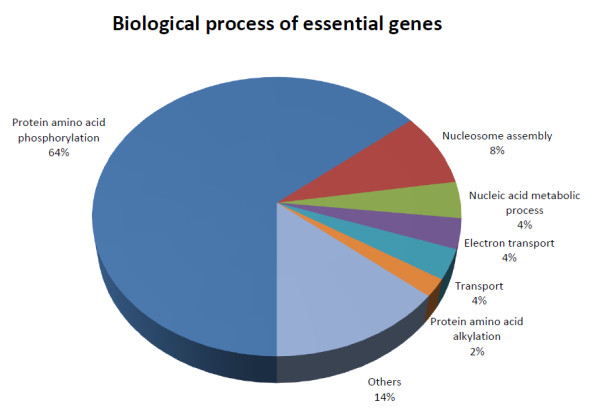
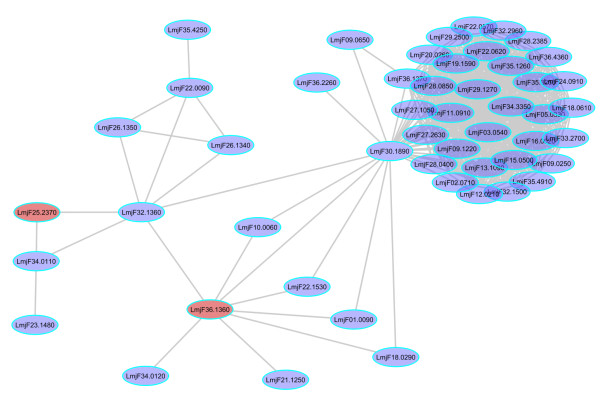
We have predicted the protein interaction network of Leishmania major by using three validated methods: PSIMAP, PEIMAP, and iPfam. Combining the results from these methods, we calculated a high confidence network (confidence score > 0.70) with 1,366 nodes and 33,861 interactions. We were able to predict the biological process for 263 interacting proteins by doing enrichment analysis of the clusters detected. Analyzing the topology of the network with metrics such as connectivity and betweenness centrality, we detected 142 potential drug targets after homology filtering with the human proteome. Further experiments can be done to validate these targets.
We have constructed the first protein interaction network of the Leishmania major parasite by using a computational approach. The topological analysis of the protein network enabled us to identify a set of candidate proteins that may be both (1) essential for parasite survival and (2) without human orthologs. These potential targets are promising for further experimental validation. This strategy, if validated, may augment established drug discovery methodologies, for this and possibly other tropical diseases, with a relatively low additional investment of time and resources.
利什曼病是一种烈性寄生虫感染,在全球范围内造成疾病负担。大多数治疗方法都有有毒的副作用,而且由于耐药菌株的出现,疗效已经下降。由于缺乏针对这种疾病的有前途的药物靶点,情况变得更加恶化。我们采用计算方法来检测新的药物靶点,这可能成为发现这种热带疾病新药的有效策略。
我们使用三种经过验证的方法 PSIMAP、PEIMAP 和 iPfam 来预测 L. major 的蛋白质相互作用网络。将这些方法的结果结合起来,我们计算了一个高置信度网络(置信度得分>0.70),其中包含 1366 个节点和 33861 个相互作用。通过对检测到的簇进行富集分析,我们能够预测 263 个相互作用蛋白的生物学过程。通过分析网络的拓扑结构,如连通性和中间中心性等指标,我们在与人类蛋白质组进行同源性过滤后,检测到 142 个潜在的药物靶点。可以进一步进行实验来验证这些靶点。
我们通过计算方法构建了第一个 L. major 寄生虫的蛋白质相互作用网络。蛋白质网络的拓扑分析使我们能够确定一组候选蛋白,这些蛋白可能(1)对寄生虫的生存至关重要,(2)没有人类同源物。这些潜在的靶标可能是进一步实验验证的良好候选物。如果得到验证,这种策略可能会为这种热带疾病甚至其他热带疾病的现有药物发现方法提供补充,只需要相对较少的额外时间和资源投入。