Department of Electrical Engineering and Computer Science and Graduate School of Genome Science and Techonology, University of Tennessee, Knoxville, TN 37996, USA.
BMC Bioinformatics. 2010 Oct 7;11 Suppl 6(Suppl 6):S14. doi: 10.1186/1471-2105-11-S6-S14.
Searching the enormous amount of information available in biomedical literature to extract novel functional relationships among genes remains a challenge in the field of bioinformatics. While numerous (software) tools have been developed to extract and identify gene relationships from biological databases, few effectively deal with extracting new (or implied) gene relationships, a process which is useful in interpretation of discovery-oriented genome-wide experiments.
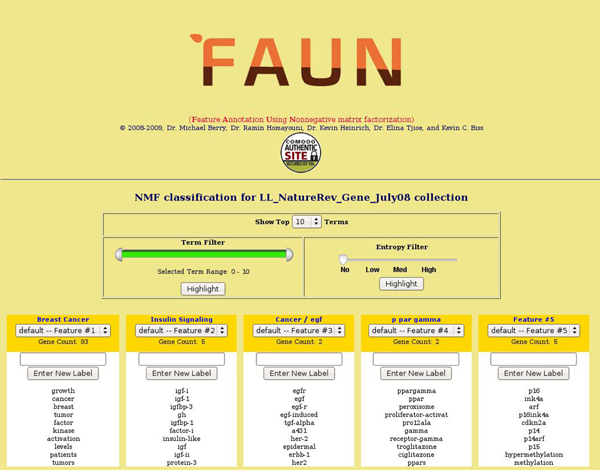
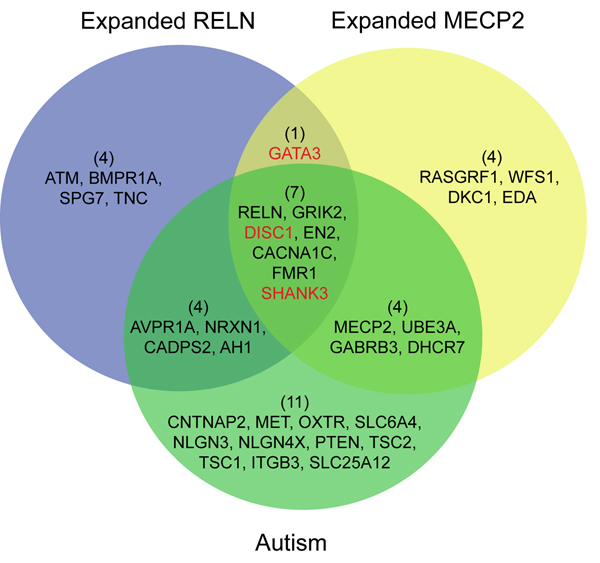
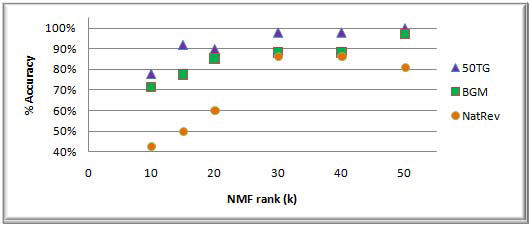
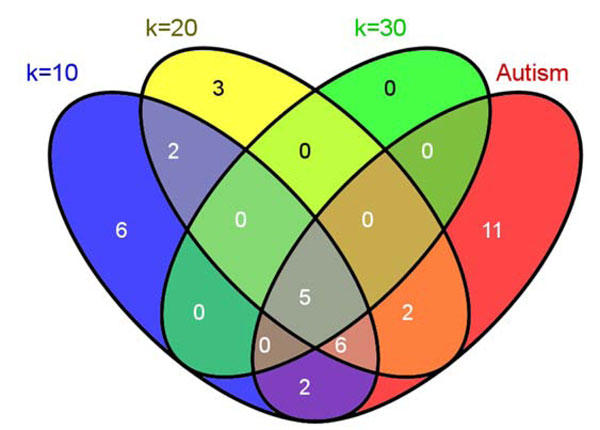
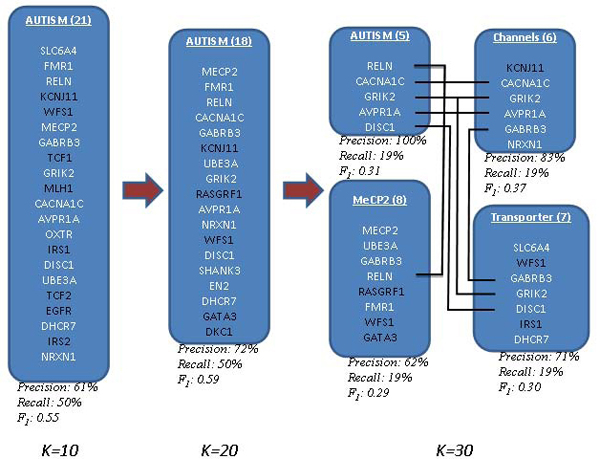
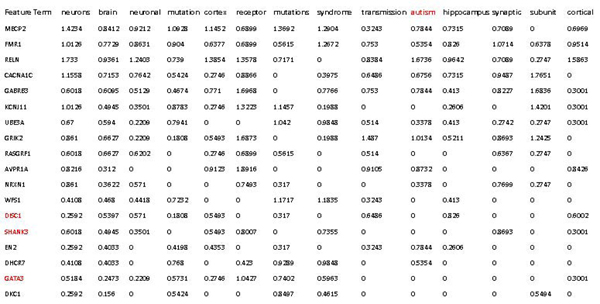
In this study, we develop a Web-based bioinformatics software environment called FAUN or Feature Annotation Using Nonnegative matrix factorization (NMF) to facilitate both the discovery and classification of functional relationships among genes. Both the computational complexity and parameterization of NMF for processing gene sets are discussed. FAUN is tested on three manually constructed gene document collections. Its utility and performance as a knowledge discovery tool is demonstrated using a set of genes associated with Autism.
FAUN not only assists researchers to use biomedical literature efficiently, but also provides utilities for knowledge discovery. This Web-based software environment may be useful for the validation and analysis of functional associations in gene subsets identified by high-throughput experiments.
在生物信息学领域,从大量生物医学文献中搜索信息以提取基因之间新的功能关系仍然是一个挑战。虽然已经开发了许多(软件)工具来从生物数据库中提取和识别基因关系,但很少有工具能够有效地提取新的(或隐含的)基因关系,而这一过程对于解释以发现为导向的全基因组实验非常有用。
在这项研究中,我们开发了一个名为 FAUN 的基于网络的生物信息学软件环境,即使用非负矩阵分解(NMF)进行特征注释,以促进基因之间功能关系的发现和分类。讨论了 NMF 处理基因集的计算复杂度和参数化。FAUN 在三个手动构建的基因文档集合上进行了测试。使用一组与自闭症相关的基因,展示了 FAUN 作为知识发现工具的实用性和性能。
FAUN 不仅可以帮助研究人员有效地利用生物医学文献,还可以提供知识发现的实用工具。这个基于网络的软件环境可能对验证和分析高通量实验中确定的基因子集的功能关联有用。