Jia Zhilong, Zhang Xiang, Guan Naiyang, Bo Xiaochen, Barnes Michael R, Luo Zhigang
Department of Chemistry and Biology, College of Science, National University of Defense Technology, Changsha, Hunan, P.R. China; William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, London, United Kingdom.
Science and Technology on Parallel and Distributed Processing Laboratory, College of Computer, National University of Defense Technology, Changsha, Hunan, P.R. China.
PLoS One. 2015 Sep 8;10(9):e0137782. doi: 10.1371/journal.pone.0137782. eCollection 2015.
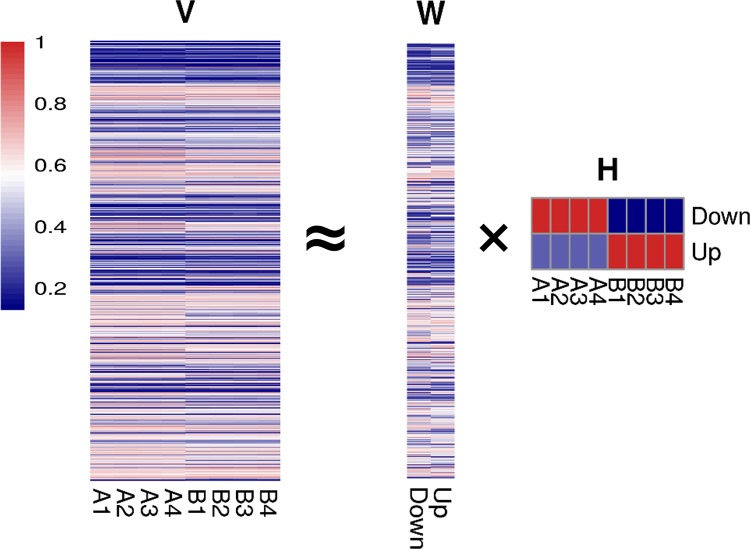
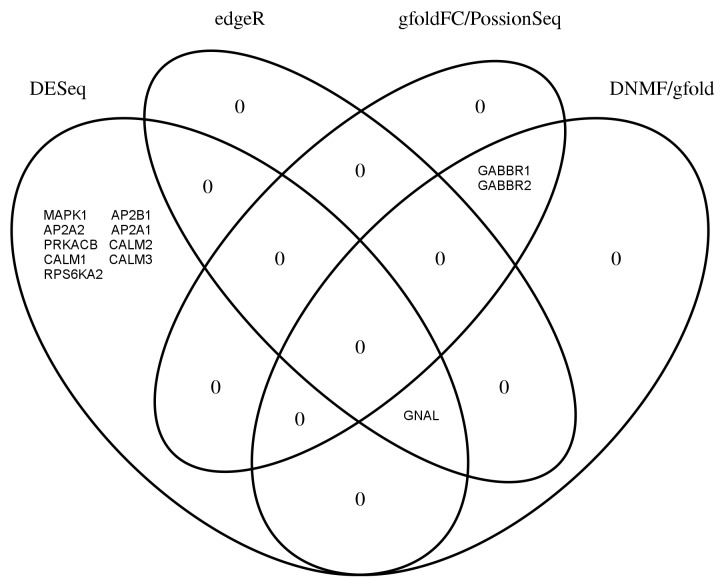
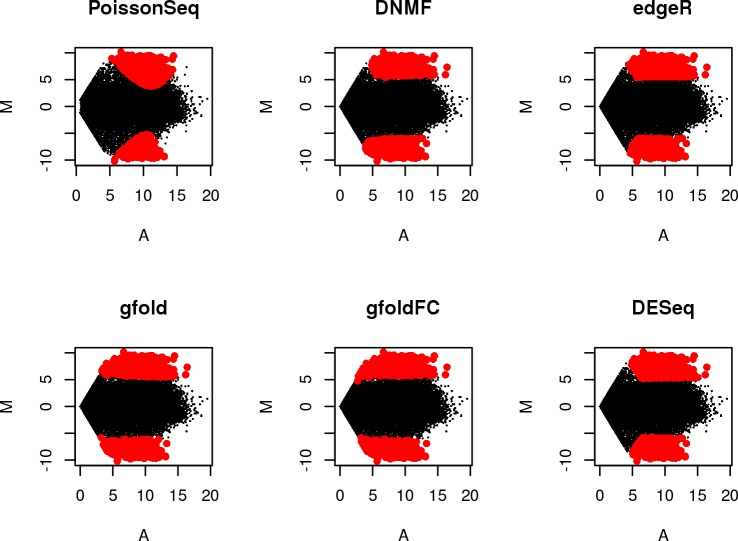
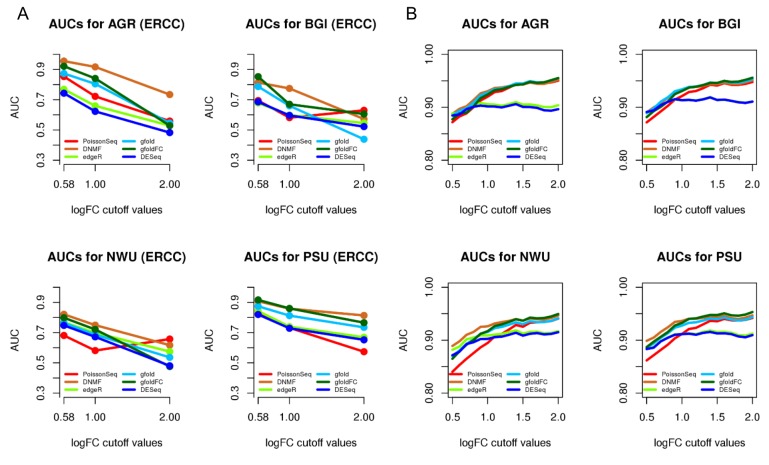
RNA-sequencing is rapidly becoming the method of choice for studying the full complexity of transcriptomes, however with increasing dimensionality, accurate gene ranking is becoming increasingly challenging. This paper proposes an accurate and sensitive gene ranking method that implements discriminant non-negative matrix factorization (DNMF) for RNA-seq data. To the best of our knowledge, this is the first work to explore the utility of DNMF for gene ranking. When incorporating Fisher's discriminant criteria and setting the reduced dimension as two, DNMF learns two factors to approximate the original gene expression data, abstracting the up-regulated or down-regulated metagene by using the sample label information. The first factor denotes all the genes' weights of two metagenes as the additive combination of all genes, while the second learned factor represents the expression values of two metagenes. In the gene ranking stage, all the genes are ranked as a descending sequence according to the differential values of the metagene weights. Leveraging the nature of NMF and Fisher's criterion, DNMF can robustly boost the gene ranking performance. The Area Under the Curve analysis of differential expression analysis on two benchmarking tests of four RNA-seq data sets with similar phenotypes showed that our proposed DNMF-based gene ranking method outperforms other widely used methods. Moreover, the Gene Set Enrichment Analysis also showed DNMF outweighs others. DNMF is also computationally efficient, substantially outperforming all other benchmarked methods. Consequently, we suggest DNMF is an effective method for the analysis of differential gene expression and gene ranking for RNA-seq data.
RNA测序正迅速成为研究转录组完整复杂性的首选方法,然而随着维度的增加,准确的基因排名变得越来越具有挑战性。本文提出了一种准确且灵敏的基因排名方法,该方法对RNA测序数据实施判别非负矩阵分解(DNMF)。据我们所知,这是探索DNMF用于基因排名效用的第一项工作。当纳入费舍尔判别准则并将降维设置为二维时,DNMF学习两个因子来近似原始基因表达数据,通过使用样本标签信息提取上调或下调的元基因。第一个因子表示两个元基因的所有基因权重,作为所有基因的加性组合,而第二个学习到的因子代表两个元基因的表达值。在基因排名阶段,所有基因根据元基因权重的差异值按降序排列。利用非负矩阵分解的性质和费舍尔准则,DNMF能够稳健地提升基因排名性能。对具有相似表型的四个RNA测序数据集的两个基准测试进行差异表达分析的曲线下面积分析表明,我们提出的基于DNMF的基因排名方法优于其他广泛使用的方法。此外,基因集富集分析也表明DNMF优于其他方法。DNMF在计算上也很高效,大大优于所有其他基准方法。因此,我们认为DNMF是一种用于分析RNA测序数据中差异基因表达和基因排名的有效方法。