BioMaPS Institute for Quantitative Biology, Rutgers University, Piscataway, New Jersey, USA.
BMC Bioinformatics. 2010 Oct 14;11:515. doi: 10.1186/1471-2105-11-515.
Comprehensively understanding corticosteroid pharmacogenomic effects is an essential step towards an insight into the underlying molecular mechanisms for both beneficial and detrimental clinical effects. Nevertheless, even in a single tissue different methods of corticosteroid administration can induce different patterns of expression and regulatory control structures. Therefore, rich in vivo datasets of pharmacological time-series with two dosing regimens sampled from rat liver are examined for temporal patterns of changes in gene expression and their regulatory commonalities.
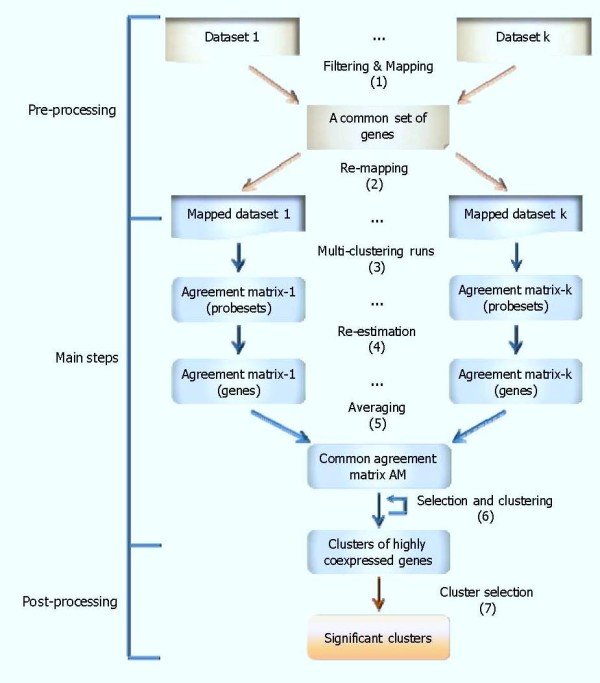
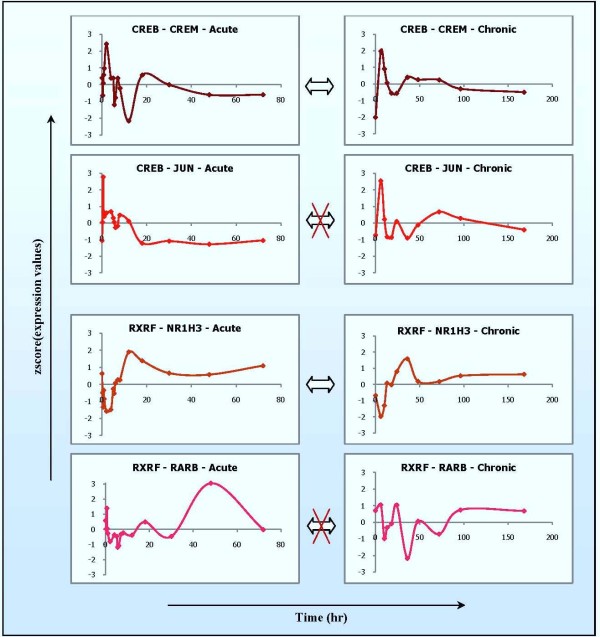
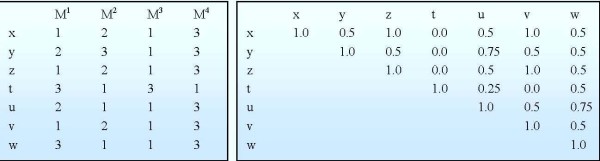
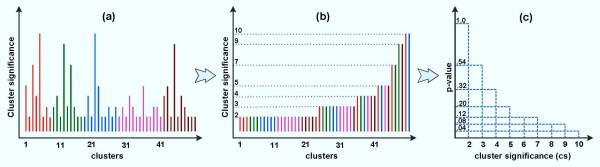
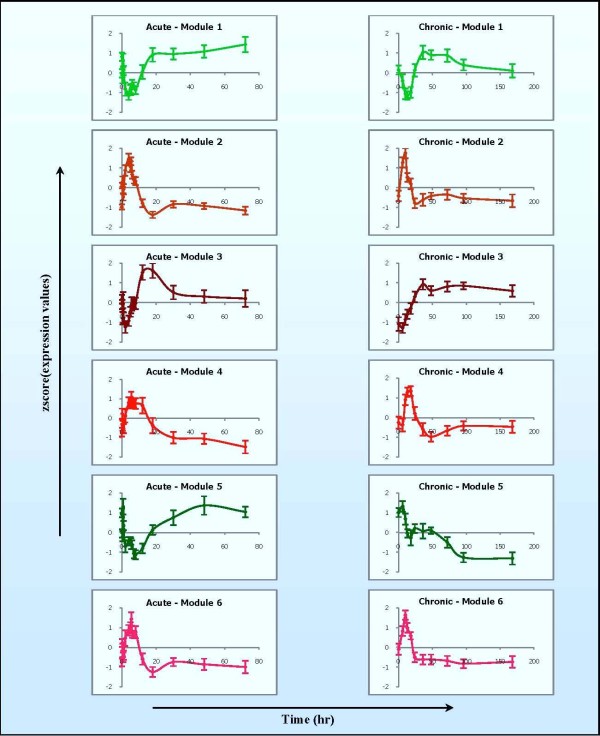
The study addresses two issues, including (1) identifying significant transcriptional modules coupled with dynamic expression patterns and (2) predicting relevant common transcriptional controls to better understand the underlying mechanisms of corticosteroid adverse effects. Following the orientation of meta-analysis, an extended computational approach that explores the concept of agreement matrix from consensus clustering has been proposed with the aims of identifying gene clusters that share common expression patterns across multiple dosing regimens as well as handling challenges in the analysis of microarray data from heterogeneous sources, e.g. different platforms and time-grids in this study. Six significant transcriptional modules coupled with typical patterns of expression have been identified. Functional analysis reveals that virtually all enriched functions (gene ontologies, pathways) in these modules are shown to be related to metabolic processes, implying the importance of these modules in adverse effects under the administration of corticosteroids. Relevant putative transcriptional regulators (e.g. RXRF, FKHD, SP1F) are also predicted to provide another source of information towards better understanding the complexities of expression patterns and the underlying regulatory mechanisms of those modules.
We have proposed a framework to identify significant coexpressed clusters of genes across multiple conditions experimented from different microarray platforms, time-grids, and also tissues if applicable. Analysis on rich in vivo datasets of corticosteroid time-series yielded significant insights into the pharmacogenomic effects of corticosteroids, especially the relevance to metabolic side-effects. This has been illustrated through enriched metabolic functions in those transcriptional modules and the presence of GRE binding motifs in those enriched pathways, providing significant modules for further analysis on pharmacogenomic corticosteroid effects.
全面了解皮质甾类药物的遗传药理学效应是深入了解其有益和有害临床效应的潜在分子机制的重要步骤。然而,即使在单一组织中,不同的皮质甾类药物给药方法也会诱导不同的表达模式和调节控制结构。因此,本研究对大鼠肝脏中两种给药方案的药理学时间序列的丰富体内数据集进行了检查,以研究基因表达的时间变化模式及其调节的共同性。
该研究解决了两个问题,包括(1)确定与动态表达模式相关的显著转录模块,以及(2)预测相关的共同转录控制,以更好地理解皮质甾类药物不良反应的潜在机制。本研究采用扩展的计算方法,从共识聚类的协议矩阵角度出发,提出了一种扩展的计算方法,旨在识别在多个给药方案中具有共同表达模式的基因簇,以及处理来自异质来源的微阵列数据分析中的挑战,例如本研究中的不同平台和时间网格。已经确定了六个与典型表达模式相关的显著转录模块。功能分析表明,这些模块中几乎所有富集的功能(基因本体论、途径)都与代谢过程有关,这意味着这些模块在皮质甾类药物给药下的不良反应中非常重要。还预测了相关的潜在转录调节剂(例如 RXRF、FKHD、SP1F),为更好地理解这些模块的表达模式和潜在调控机制提供了另一种信息来源。
我们提出了一种框架,用于识别来自不同微阵列平台、时间网格甚至组织(如果适用)的多个条件下的显著共表达基因簇。对皮质甾类药物时间序列的丰富体内数据集进行分析,深入了解了皮质甾类药物的遗传药理学效应,特别是与代谢副作用的相关性。这通过这些转录模块中丰富的代谢功能和这些富集途径中 GRE 结合基序的存在得到了说明,为进一步分析皮质甾类药物的遗传药理学效应提供了重要的模块。