Institute of Evolutionary Biology, University of Edinburgh, West Mains Road, Edinburgh EH9 3JT, UK.
BMC Genomics. 2010 Oct 16;11:571. doi: 10.1186/1471-2164-11-571.
Roche 454 pyrosequencing has become a method of choice for generating transcriptome data from non-model organisms. Once the tens to hundreds of thousands of short (250-450 base) reads have been produced, it is important to correctly assemble these to estimate the sequence of all the transcripts. Most transcriptome assembly projects use only one program for assembling 454 pyrosequencing reads, but there is no evidence that the programs used to date are optimal. We have carried out a systematic comparison of five assemblers (CAP3, MIRA, Newbler, SeqMan and CLC) to establish best practices for transcriptome assemblies, using a new dataset from the parasitic nematode Litomosoides sigmodontis.
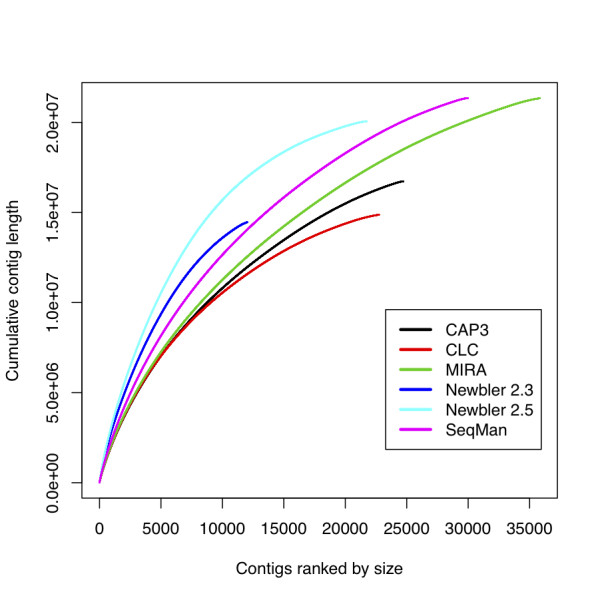
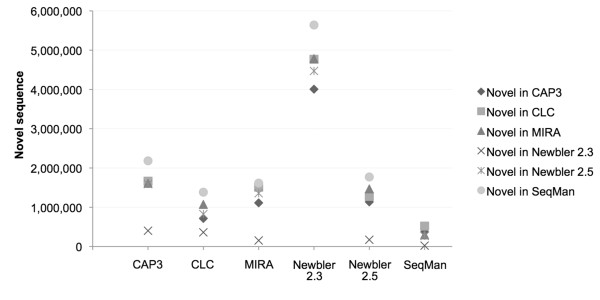
Although no single assembler performed best on all our criteria, Newbler 2.5 gave longer contigs, better alignments to some reference sequences, and was fast and easy to use. SeqMan assemblies performed best on the criterion of recapitulating known transcripts, and had more novel sequence than the other assemblers, but generated an excess of small, redundant contigs. The remaining assemblers all performed almost as well, with the exception of Newbler 2.3 (the version currently used by most assembly projects), which generated assemblies that had significantly lower total length. As different assemblers use different underlying algorithms to generate contigs, we also explored merging of assemblies and found that the merged datasets not only aligned better to reference sequences than individual assemblies, but were also more consistent in the number and size of contigs.
Transcriptome assemblies are smaller than genome assemblies and thus should be more computationally tractable, but are often harder because individual contigs can have highly variable read coverage. Comparing single assemblers, Newbler 2.5 performed best on our trial data set, but other assemblers were closely comparable. Combining differently optimal assemblies from different programs however gave a more credible final product, and this strategy is recommended.
罗氏 454 焦磷酸测序已成为从非模式生物中生成转录组数据的首选方法。一旦产生了数万到数十万条短(250-450 个碱基)的读取序列,正确组装这些序列以估计所有转录本的序列就变得非常重要。大多数转录组组装项目仅使用一个程序来组装 454 焦磷酸测序读取序列,但迄今为止没有证据表明使用的程序是最优的。我们使用来自寄生线虫旋毛虫的新数据集,对五个组装程序(CAP3、MIRA、Newbler、SeqMan 和 CLC)进行了系统比较,以确定转录组组装的最佳实践。
尽管没有一个程序在我们的所有标准上都表现最好,但 Newbler 2.5 生成的序列更长,与一些参考序列的比对更好,并且使用快速且简单。SeqMan 组装在重现已知转录本的标准上表现最好,并且比其他组装程序具有更多的新序列,但生成的小冗余序列过多。其余的组装程序表现几乎相同,除了 Newbler 2.3(目前大多数组装项目使用的版本),它生成的组装序列总长度明显较低。由于不同的组装程序使用不同的底层算法来生成序列,我们还探索了组装的合并,发现合并后的数据集不仅比对参考序列的对齐更好,而且在序列的数量和大小上也更一致。
转录组组装比基因组组装小,因此应该更容易进行计算,但通常更难,因为单个序列可能具有高度可变的读取覆盖度。在我们的试验数据集上比较单个组装程序时,Newbler 2.5 表现最佳,但其他组装程序也非常接近。然而,从不同的程序中组合不同的最佳组装程序会得到更可信的最终产品,因此推荐使用这种策略。