Graduate Institute of Statistics and Information Science, National Changhua University of Education, No.1 Jinde Road, Changhua City, Changhua County, 50007, Taiwan.
Institute of Statistics, National Chiao Tung University, 1001 University Road, Hsinchu, 30010, Taiwan.
Sci Rep. 2020 Jun 26;10(1):10493. doi: 10.1038/s41598-020-64353-1.
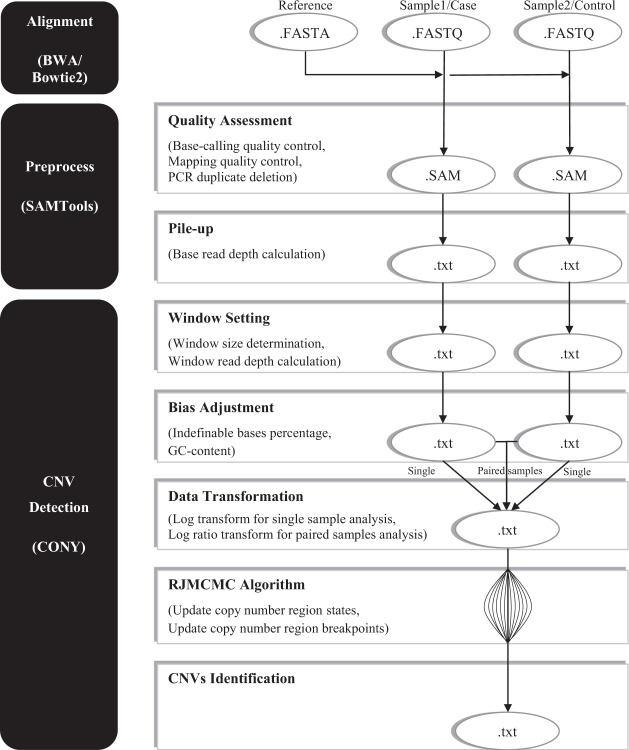
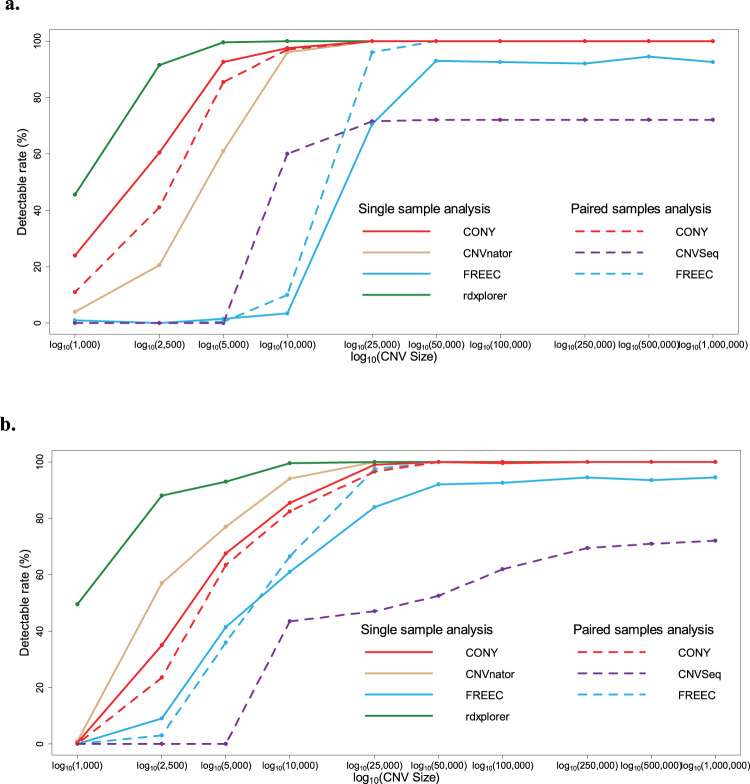
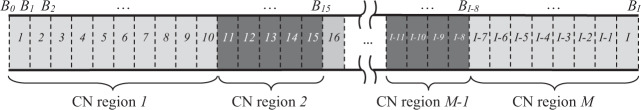
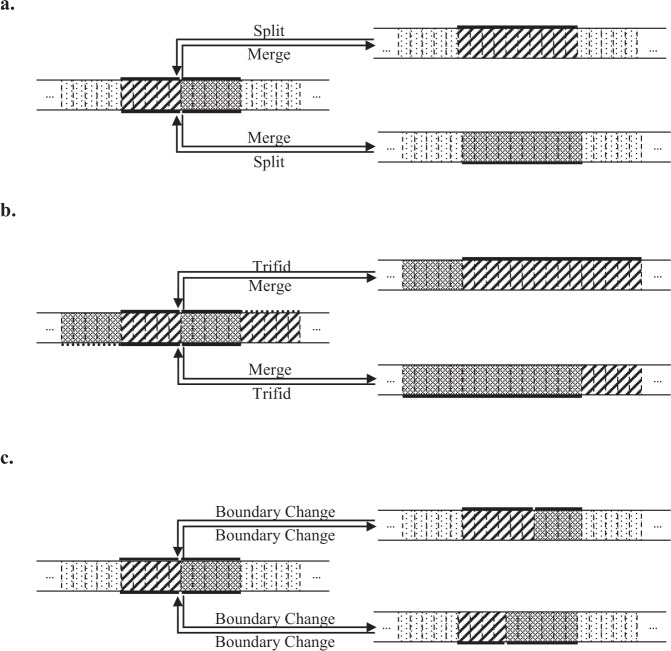
Copy number variations (CNVs) are genomic structural mutations consisting of abnormal numbers of fragment copies. Next-generation sequencing of read-depth signals mirrors these variants. Some tools used to predict CNVs by depth have been published, but most of these tools can be applied to only a specific data type due to modeling limitations. We develop a tool for copy number variation detection by a Bayesian procedure, i.e., CONY, that adopts a Bayesian hierarchical model and an efficient reversible-jump Markov chain Monte Carlo inference algorithm for whole genome sequencing of read-depth data. CONY can be applied not only to individual samples for estimating the absolute number of copies but also to case-control pairs for detecting patient-specific variations. We evaluate the performance of CONY and compare CONY with competing approaches through simulations and by using experimental data from the 1000 Genomes Project. CONY outperforms the other methods in terms of accuracy in both single-sample and paired-samples analyses. In addition, CONY performs well regardless of whether the data coverage is high or low. CONY is useful for detecting both absolute and relative CNVs from read-depth data sequences. The package is available at https://github.com/weiyuchung/CONY.
拷贝数变异(CNVs)是由片段拷贝数异常引起的基因组结构突变。新一代测序的读深信号反映了这些变体。已经发表了一些用于通过深度预测 CNVs 的工具,但由于建模限制,这些工具大多数只能应用于特定的数据类型。我们开发了一种基于贝叶斯过程的拷贝数变异检测工具,即 CONY,它采用贝叶斯层次模型和高效的可逆跳跃马尔可夫链蒙特卡罗推断算法,用于对读深数据进行全基因组测序。CONY 不仅可以应用于个体样本,以估计绝对拷贝数,还可以应用于病例对照对,以检测患者特异性变异。我们通过模拟和使用来自 1000 基因组计划的实验数据来评估 CONY 的性能,并将 CONY 与竞争方法进行比较。CONY 在单样本和配对样本分析中的准确性都优于其他方法。此外,无论数据覆盖率高低,CONY 的性能都很好。CONY 可用于从读深数据序列中检测绝对和相对 CNVs。该软件包可在 https://github.com/weiyuchung/CONY 获得。