Department of Immunology and Infectious Diseases, Harvard School of Public Health, Boston, MA, USA.
BMC Genomics. 2013 Mar 5;14:145. doi: 10.1186/1471-2164-14-145.
Mycobacterial interspersed repetitive units (MIRUs) are minisatellites within the Mycobacterium tuberculosis (Mtb) genome. Copy number variation (CNV) in MIRU loci is used for epidemiological typing, making the rate of variation important for tracking the transmission of Mtb strains. In this study, we developed and assessed a whole-genome sequencing (WGS) approach to detect MIRU CNV in Mtb. We applied this methodology to a panel of Mtb strains isolated from the macaque model of tuberculosis (TB), the animal model that best mimics human disease. From these data, we have estimated the rate of MIRU variation in the host environment, providing a benchmark rate for future epidemiologic work.
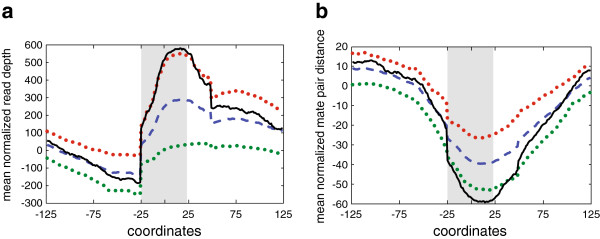
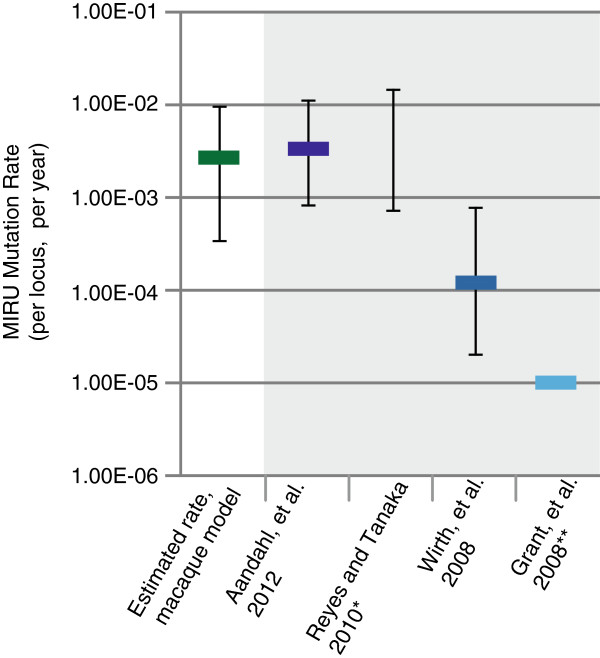
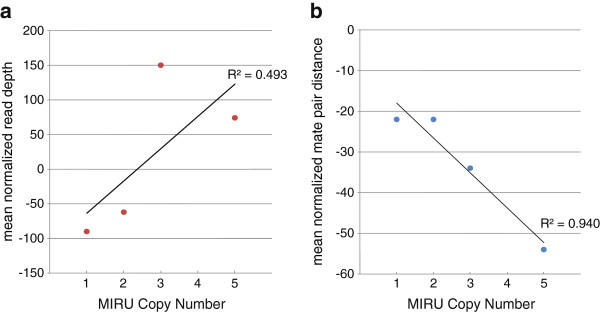
We assessed variation at the 24 MIRU loci used for typing in a set of Mtb strains isolated from infected cynomolgus macaques. We previously performed WGS of these strains and here have applied both read depth (RD) and paired-end mapping (PEM) metrics to identify putative copy number variants. To assess the relative power of these approaches, all MIRU loci were resequenced using Sanger sequencing. We detected two insertion/deletion events both of which could be identified as candidates by PEM criteria. With these data, we estimate a MIRU mutation rate of 2.70 × 10-03 (95% CI: 3.30 × 10-04- 9.80 × 10-03) per locus, per year.
Our results represent the first experimental estimate of the MIRU mutation rate in Mtb. This rate is comparable to the highest previous estimates gathered from epidemiologic data and meta-analyses. Our findings allow for a more rigorous interpretation of data gathered from MIRU typing.
分枝杆菌散布重复单位(MIRUs)是分枝杆菌结核(Mtb)基因组内的微卫星。MIRU 基因座的拷贝数变异(CNV)用于流行病学分型,因此变异率对于追踪 Mtb 菌株的传播非常重要。在这项研究中,我们开发并评估了一种全基因组测序(WGS)方法来检测 Mtb 中的 MIRU CNV。我们将这种方法应用于从结核猴模型中分离的 Mtb 菌株的一组面板中,该模型是最能模拟人类疾病的动物模型。从这些数据中,我们估计了宿主环境中 MIRU 变异的速度,为未来的流行病学工作提供了基准速度。
我们评估了一组从感染食蟹猴的 Mtb 菌株中分离的菌株中用于分型的 24 个 MIRU 基因座的变异情况。我们之前对这些菌株进行了 WGS 分析,并且在这里应用了读深度(RD)和配对末端映射(PEM)指标来识别潜在的拷贝数变异。为了评估这些方法的相对能力,我们使用 Sanger 测序对所有 MIRU 基因座进行了重新测序。我们检测到两个插入/缺失事件,其中两个都可以通过 PEM 标准被识别为候选事件。根据这些数据,我们估计每个 MIRU 基因座每年的突变率为 2.70×10-03(95%CI:3.30×10-04-9.80×10-03)。
我们的结果代表了 Mtb 中 MIRU 突变率的首次实验估计。这个速度与从流行病学数据和荟萃分析中获得的最高先前估计值相当。我们的发现使得可以更严格地解释从 MIRU 分型中获得的数据。