Plant Stress Genomics Research Center, Division of Chemical and Life Sciences and Engineering, King Abdullah University of Science and Technology, Thuwal 23955-6900, Kingdom of Saudi Arabia.
Biol Direct. 2010 Nov 8;5:63. doi: 10.1186/1745-6150-5-63.
Understanding the compositional dynamics of genomes and their coding sequences is of great significance in gaining clues into molecular evolution and a large number of publically-available genome sequences have allowed us to quantitatively predict deviations of empirical data from their theoretical counterparts. However, the quantification of theoretical compositional variations for a wide diversity of genomes remains a major challenge.
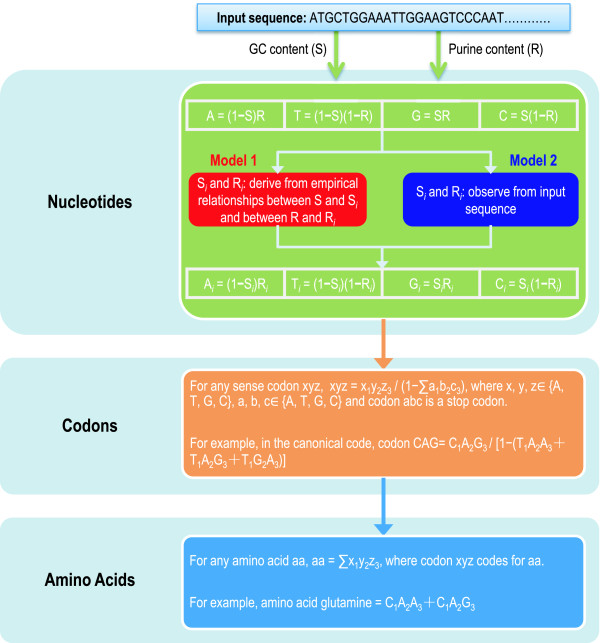
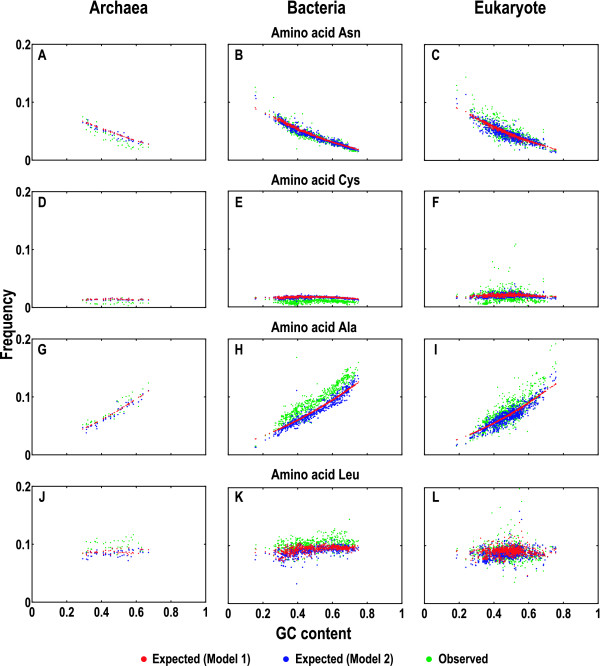
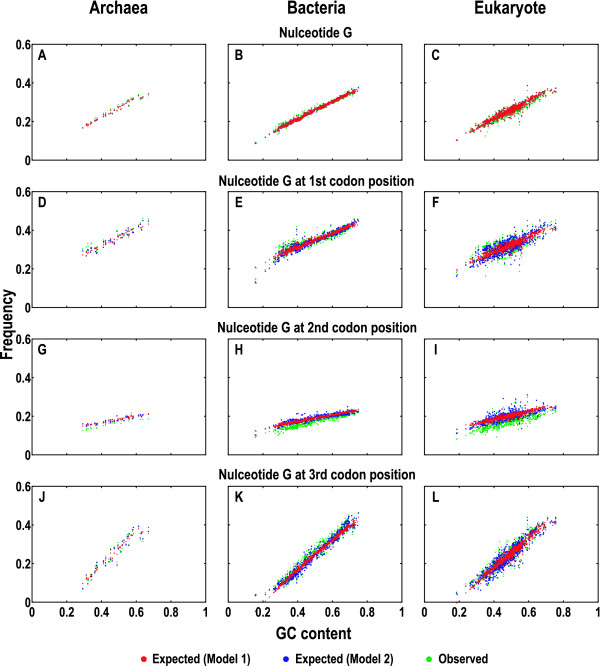
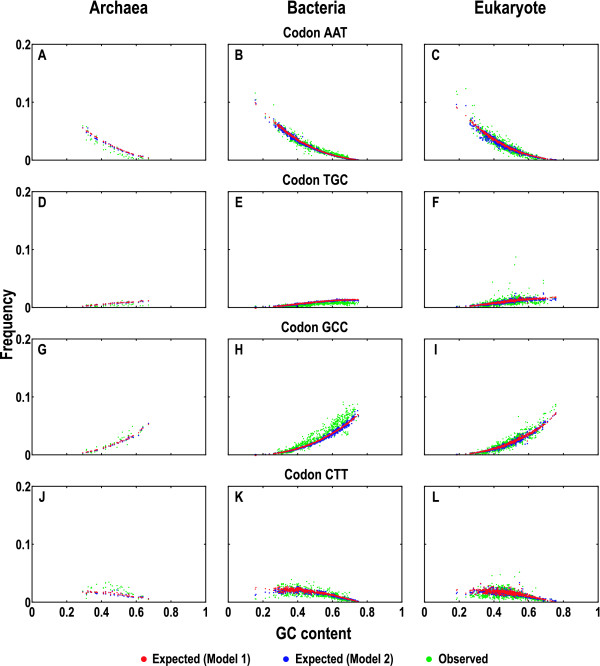
To model the compositional dynamics of protein-coding sequences, we propose two simple models that take into account both mutation and selection effects, which act differently at the three codon positions, and use both GC and purine contents as compositional parameters. The two models concern the theoretical composition of nucleotides, codons, and amino acids, with no prerequisite of homologous sequences or their alignments. We evaluated the two models by quantifying theoretical compositions of a large collection of protein-coding sequences (including 46 of Archaea, 686 of Bacteria, and 826 of Eukarya), yielding consistent theoretical compositions across all the collected sequences.
We show that the compositions of nucleotides, codons, and amino acids are largely determined by both GC and purine contents and suggest that deviations of the observed from the expected compositions may reflect compositional signatures that arise from a complex interplay between mutation and selection via DNA replication and repair mechanisms.
理解基因组及其编码序列的组成动态对于揭示分子进化的线索具有重要意义,并且大量公开的基因组序列使我们能够定量预测经验数据与理论数据之间的偏差。然而,对各种基因组的理论组成变化进行量化仍然是一个主要挑战。
为了模拟蛋白质编码序列的组成动态,我们提出了两个简单的模型,这些模型考虑了突变和选择的影响,这些影响在三个密码子位置上的作用不同,并将 GC 和嘌呤含量用作组成参数。这两个模型涉及核苷酸、密码子和氨基酸的理论组成,不需要同源序列或其比对。我们通过量化大量蛋白质编码序列(包括 46 个古细菌、686 个细菌和 826 个真核生物)的理论组成来评估这两个模型,从而在所有收集的序列中产生一致的理论组成。
我们表明,核苷酸、密码子和氨基酸的组成主要由 GC 和嘌呤含量决定,并表明观察到的组成与预期组成的偏差可能反映了由 DNA 复制和修复机制介导的突变和选择之间复杂相互作用产生的组成特征。