Lin Y-X, Baladandayuthapani V, Bonato V, Do K-A
Centre for Statistical and Survey Methodology, School of Mathematics and Applied Statistics, University of Wollongong NSW 2522, Australia.
Cancer Inform. 2010 Oct 12;9:229-49. doi: 10.4137/CIN.S5614.
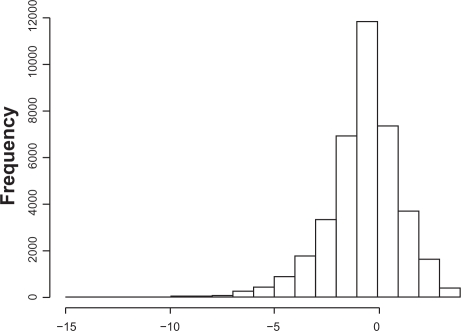
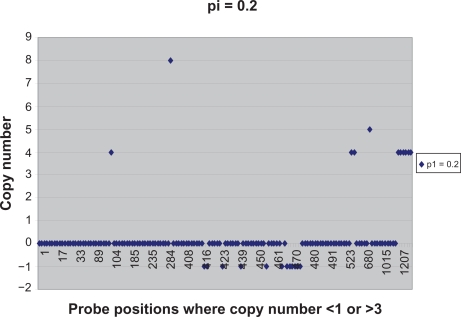
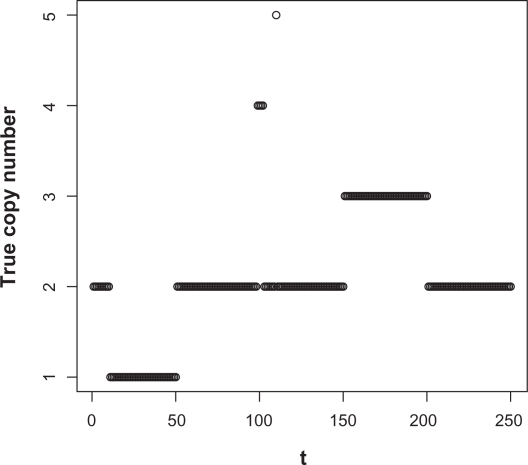
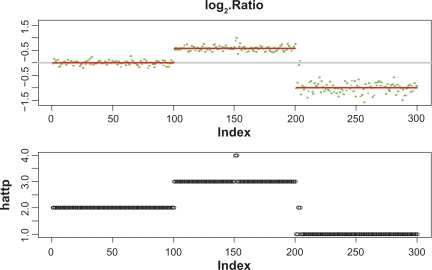
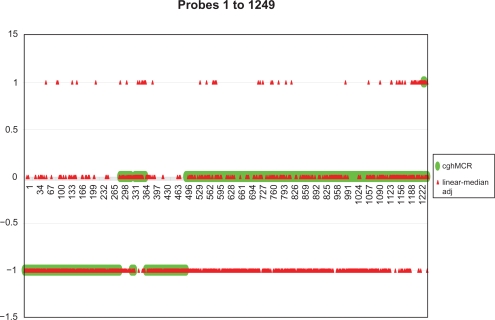
Existing methods for estimating copy number variations in array comparative genomic hybridization (aCGH) data are limited to estimations of the gain/loss of chromosome regions for single sample analysis. We propose the linear-median method for estimating shared copy numbers in DNA sequences across multiple samples, demonstrate its operating characteristics through simulations and applications to real cancer data, and compare it to two existing methods.
Our proposed linear-median method has the power to estimate common changes that appear at isolated single probe positions or very short regions. Such changes are hard to detect by current methods. This new method shows a higher rate of true positives and a lower rate of false positives. The linear-median method is non-parametric and hence is more robust in estimating copy number. Additionally the linear-median method is easily computable for practical aCGH data sets compared to other copy number estimation methods.
现有用于估计阵列比较基因组杂交(aCGH)数据中拷贝数变异的方法仅限于对单个样本分析的染色体区域增益/缺失的估计。我们提出了线性中位数方法来估计多个样本DNA序列中的共享拷贝数,通过模拟和对实际癌症数据的应用展示其操作特性,并将其与两种现有方法进行比较。
我们提出的线性中位数方法有能力估计出现在孤立单探针位置或非常短区域的常见变化。这种变化目前的方法难以检测到。这种新方法显示出更高的真阳性率和更低的假阳性率。线性中位数方法是非参数的,因此在估计拷贝数时更稳健。此外,与其他拷贝数估计方法相比,线性中位数方法对于实际的aCGH数据集易于计算。