Department of Pharmaceutical Sciences, University of Connecticut, 69 N. Eagleville Rd., Storrs, CT 06269, USA.
J Mol Graph Model. 2011 Feb;29(5):608-13. doi: 10.1016/j.jmgm.2010.11.004. Epub 2010 Nov 11.
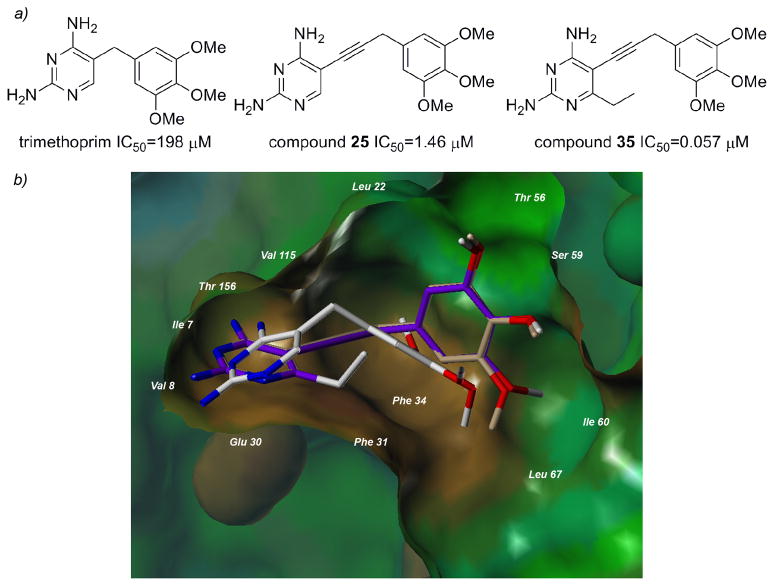
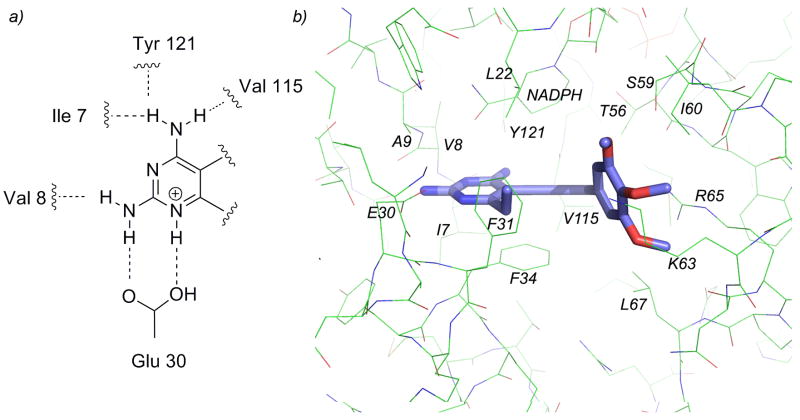
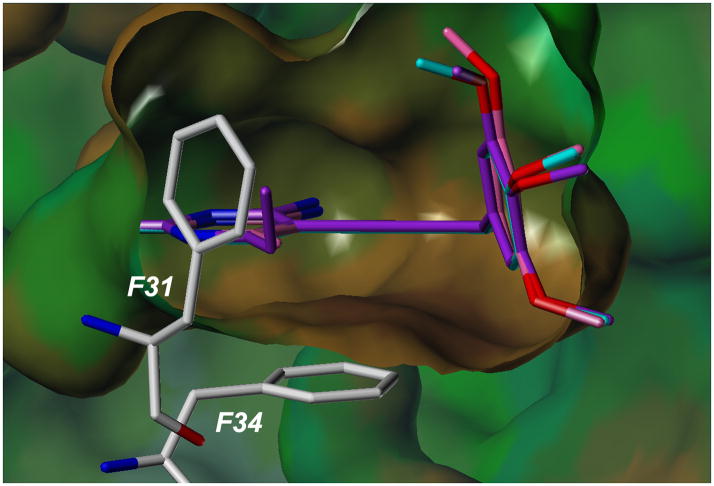
Dihydrofolate reductase (DHFR) has been a well-recognized target for the development of therapeutics for human cancers for several decades. Classical inhibitors of DHFR use an active transport mechanism to gain access to the cell; disabling this mechanism creates a pathway for resistance. In response, recent research focuses on nonclassical lipid-soluble DHFR inhibitors that are designed to passively diffuse through the membrane. Here, a new series of propargyl-linked antifolates are investigated as potential nonclassical human DHFR inhibitors. Several of these compounds exhibit potent enzyme inhibition with 50% inhibition concentration values under 500 nM. Molecular docking investigations show that the compounds maintain conserved hydrogen bonds between the pyrimidine ring and the enzyme as well as form van der Waals interactions with critical residues in the active site. Interestingly, the most potent compound, 2,4-diamino-5-(3-(3,4,5-trimethoxyphenyl)prop-1-ynyl)-6-ethylpyrimidine (compound 35), is 3500-fold more potent than trimethoprim, a potent inhibitor of bacterial DHFR but weak inhibitor of human DHFR. The two structural differences between compound 35 and trimethoprim show that the propargyl linkage and the substitution at C6 of the pyrimidine ring are critical to the formation of contacts with Thr 56, Ser 59, Ile 60, Leu 22, Phe 31 and Phe 34 and hence, to enhancing potency. The propargyl-linked antifolates are efficient ligands with a high ratio of potency to the number of non-hydrogen atoms and represent a potentially fruitful avenue for future development of antineoplastic agents.
二氢叶酸还原酶(DHFR)几十年来一直是开发人类癌症治疗药物的公认靶点。DHFR 的经典抑制剂利用主动转运机制进入细胞;这种机制的失活为耐药性创造了途径。因此,最近的研究集中在非经典脂溶性 DHFR 抑制剂上,这些抑制剂旨在通过被动扩散穿过膜。在这里,研究了一系列新的炔丙基连接的抗叶酸化合物,作为潜在的非经典人 DHFR 抑制剂。这些化合物中的几种具有很强的酶抑制作用,半数抑制浓度值低于 500 nM。分子对接研究表明,这些化合物在嘧啶环和酶之间保持保守的氢键,并与活性位点中的关键残基形成范德华相互作用。有趣的是,最有效的化合物 2,4-二氨基-5-(3-(3,4,5-三甲氧基苯基)丙-1-炔基)-6-乙基嘧啶(化合物 35)比三甲氧苄啶(trimethoprim)强 3500 倍,三甲氧苄啶是一种有效的细菌 DHFR 抑制剂,但对人 DHFR 的抑制作用较弱。化合物 35 和三甲氧苄啶之间的两个结构差异表明,炔丙基键和嘧啶环的 C6 取代对于与 Thr 56、Ser 59、Ile 60、Leu 22、Phe 31 和 Phe 34 形成接触至关重要,从而增强了效力。炔丙基连接的抗叶酸是高效配体,其效力与非氢原子数的比值很高,代表了未来抗肿瘤药物开发的潜在有前途的途径。