Department of Pathology, Stanford University School of Medicine, Stanford, California, United States of America.
PLoS Genet. 2010 Dec 9;6(12):e1001237. doi: 10.1371/journal.pgen.1001237.
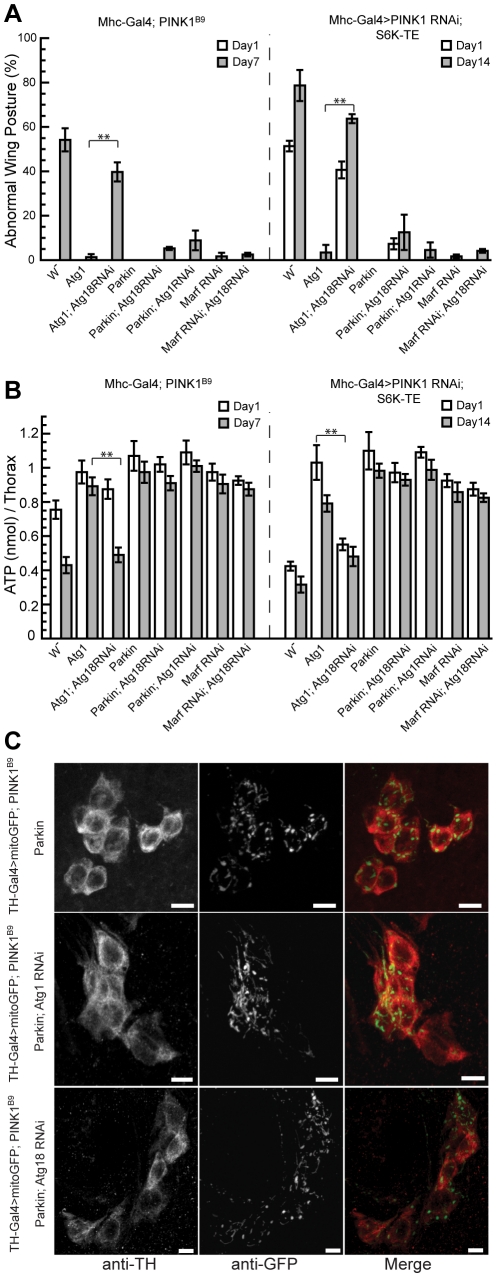
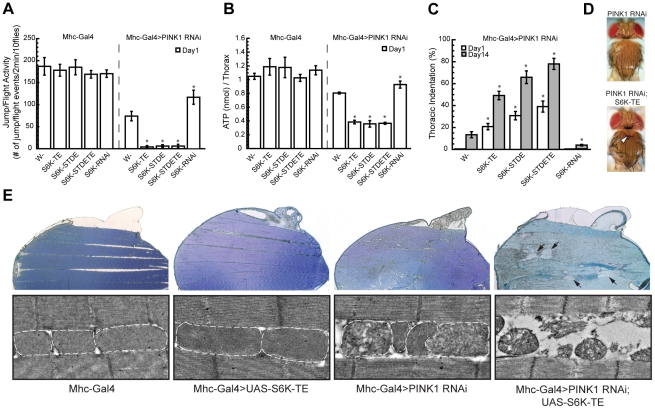
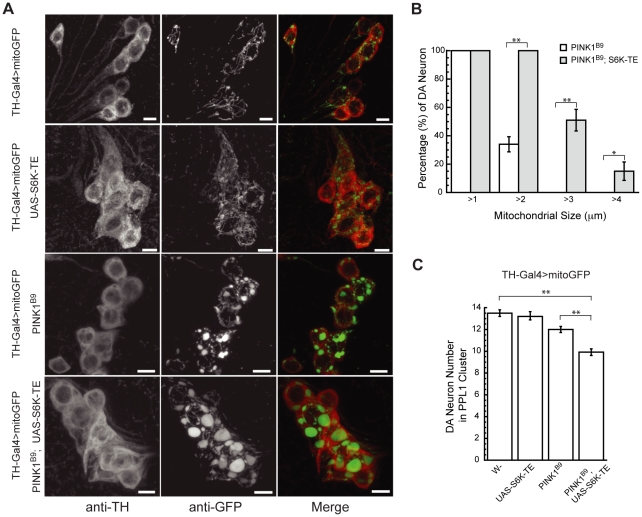
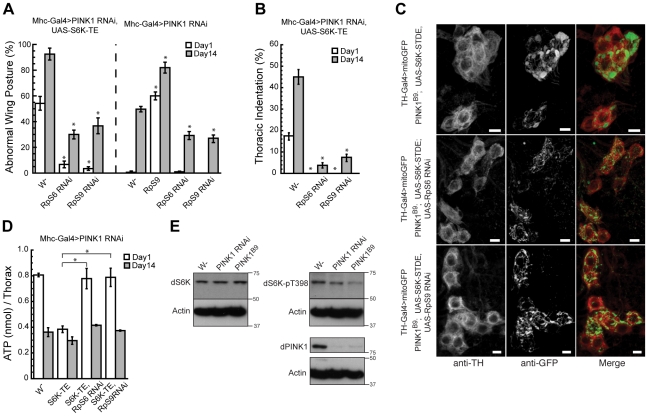
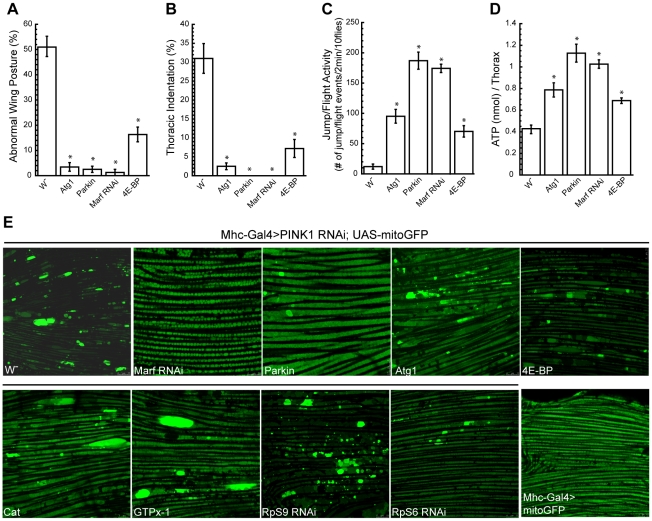
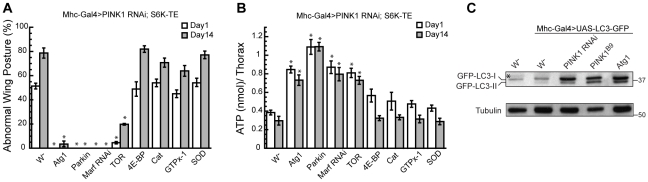
Mutations in PINK1 and Parkin cause familial, early onset Parkinson's disease. In Drosophila melanogaster, PINK1 and Parkin mutants show similar phenotypes, such as swollen and dysfunctional mitochondria, muscle degeneration, energy depletion, and dopaminergic (DA) neuron loss. We previously showed that PINK1 and Parkin genetically interact with the mitochondrial fusion/fission pathway, and PINK1 and Parkin were recently proposed to form a mitochondrial quality control system that involves mitophagy. However, the in vivo relationships among PINK1/Parkin function, mitochondrial fission/fusion, and autophagy remain unclear; and other cellular events critical for PINK1 pathogenesis remain to be identified. Here we show that PINK1 genetically interacted with the protein translation pathway. Enhanced translation through S6K activation significantly exacerbated PINK1 mutant phenotypes, whereas reduction of translation showed suppression. Induction of autophagy by Atg1 overexpression also rescued PINK1 mutant phenotypes, even in the presence of activated S6K. Downregulation of translation and activation of autophagy were already manifested in PINK1 mutant, suggesting that they represent compensatory cellular responses to mitochondrial dysfunction caused by PINK1 inactivation, presumably serving to conserve energy. Interestingly, the enhanced PINK1 mutant phenotype in the presence of activated S6K could be fully rescued by Parkin, apparently in an autophagy-independent manner. Our results reveal complex cellular responses to PINK1 inactivation and suggest novel therapeutic strategies through manipulation of the compensatory responses.
PINK1 和 Parkin 的突变会导致家族性早发性帕金森病。在黑腹果蝇中,PINK1 和 Parkin 突变体表现出相似的表型,如肿胀和功能失调的线粒体、肌肉退化、能量耗竭和多巴胺能(DA)神经元丢失。我们之前表明,PINK1 和 Parkin 与线粒体融合/裂变途径在遗传上相互作用,并且最近提出 PINK1 和 Parkin 形成了一个涉及自噬的线粒体质量控制系统。然而,PINK1/Parkin 功能、线粒体裂变/融合和自噬之间的体内关系仍不清楚;并且对于 PINK1 发病机制至关重要的其他细胞事件仍有待确定。在这里,我们表明 PINK1 与蛋白质翻译途径在遗传上相互作用。通过 S6K 激活增强翻译显著加剧了 PINK1 突变体的表型,而减少翻译则显示出抑制作用。通过 Atg1 过表达诱导自噬也挽救了 PINK1 突变体的表型,即使在激活的 S6K 存在下也是如此。在 PINK1 突变体中已经表现出翻译的下调和自噬的激活,这表明它们代表了对由 PINK1 失活引起的线粒体功能障碍的代偿性细胞反应,可能有助于节省能量。有趣的是,在激活的 S6K 存在下增强的 PINK1 突变体表型可以被 Parkin 完全挽救,显然是以一种不依赖自噬的方式。我们的结果揭示了对 PINK1 失活的复杂细胞反应,并提出了通过操纵代偿反应的新治疗策略。