Boyd Orr Centre for Population and Ecosystem Health, University of Glasgow, Glasgow, United Kingdom.
PLoS Comput Biol. 2010 Dec 9;6(12):e1001027. doi: 10.1371/journal.pcbi.1001027.
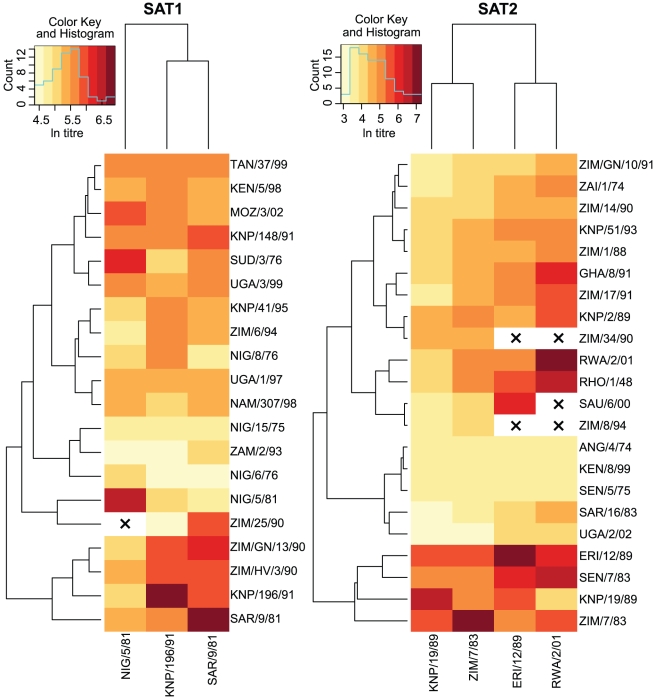
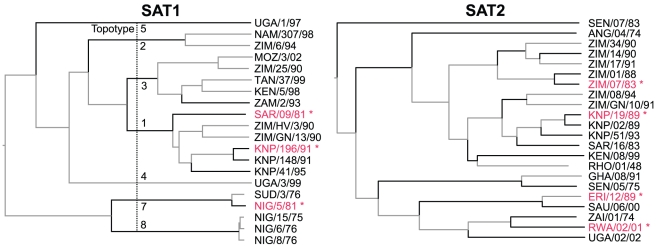
Identifying when past exposure to an infectious disease will protect against newly emerging strains is central to understanding the spread and the severity of epidemics, but the prediction of viral cross-protection remains an important unsolved problem. For foot-and-mouth disease virus (FMDV) research in particular, improved methods for predicting this cross-protection are critical for predicting the severity of outbreaks within endemic settings where multiple serotypes and subtypes commonly co-circulate, as well as for deciding whether appropriate vaccine(s) exist and how much they could mitigate the effects of any outbreak. To identify antigenic relationships and their predictors, we used linear mixed effects models to account for variation in pairwise cross-neutralization titres using only viral sequences and structural data. We identified those substitutions in surface-exposed structural proteins that are correlates of loss of cross-reactivity. These allowed prediction of both the best vaccine match for any single virus and the breadth of coverage of new vaccine candidates from their capsid sequences as effectively as or better than serology. Sub-sequences chosen by the model-building process all contained sites that are known epitopes on other serotypes. Furthermore, for the SAT1 serotype, for which epitopes have never previously been identified, we provide strong evidence--by controlling for phylogenetic structure--for the presence of three epitopes across a panel of viruses and quantify the relative significance of some individual residues in determining cross-neutralization. Identifying and quantifying the importance of sites that predict viral strain cross-reactivity not just for single viruses but across entire serotypes can help in the design of vaccines with better targeting and broader coverage. These techniques can be generalized to any infectious agents where cross-reactivity assays have been carried out. As the parameterization uses pre-existing datasets, this approach quickly and cheaply increases both our understanding of antigenic relationships and our power to control disease.
确定过去接触传染病会对新出现的菌株提供保护作用,这对于了解传染病的传播和严重程度至关重要,但预测病毒交叉保护仍然是一个重要的未解决问题。特别是对于口蹄疫病毒(FMDV)研究,改进预测这种交叉保护的方法对于预测在多种血清型和亚型共同循环的地方性流行环境中的爆发严重程度至关重要,因为这可以预测是否存在适当的疫苗以及它们可以在多大程度上减轻任何爆发的影响。为了确定抗原关系及其预测因子,我们使用线性混合效应模型,仅使用病毒序列和结构数据来解释成对交叉中和效价的变化。我们确定了在表面暴露的结构蛋白中与交叉反应性丧失相关的那些取代。这些取代可以有效地预测任何单个病毒的最佳疫苗匹配,以及新疫苗候选物的覆盖范围,与血清学一样或更好。模型构建过程中选择的子序列都包含已知在其他血清型上的表位。此外,对于 SAT1 血清型,我们从未确定过表位,我们通过控制进化结构提供了强有力的证据,证明在一组病毒中存在三个表位,并量化了一些个别残基在决定交叉中和中的相对重要性。确定和量化预测病毒株交叉反应性的位点的重要性,不仅对于单个病毒,而且对于整个血清型都很重要,可以帮助设计具有更好靶向性和更广泛覆盖范围的疫苗。这些技术可以推广到已经进行了交叉反应性测定的任何传染性病原体。由于参数化使用了现有的数据集,因此这种方法可以快速而廉价地提高我们对抗原关系的理解,并提高我们控制疾病的能力。