Institute for Molecular Medicine Finland (FIMM), University of Helsinki, Helsinki, Finland.
PLoS One. 2010 Dec 3;5(12):e15068. doi: 10.1371/journal.pone.0015068.
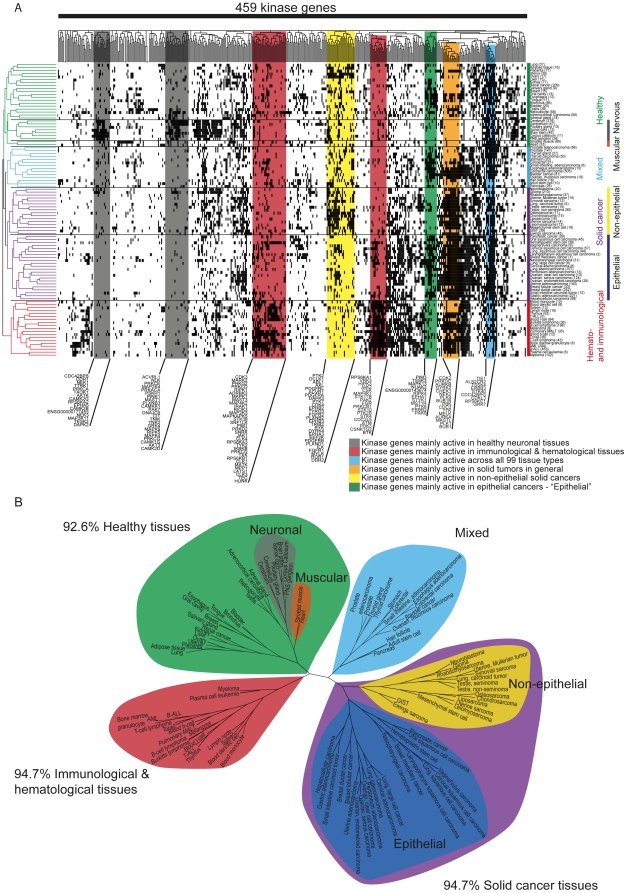
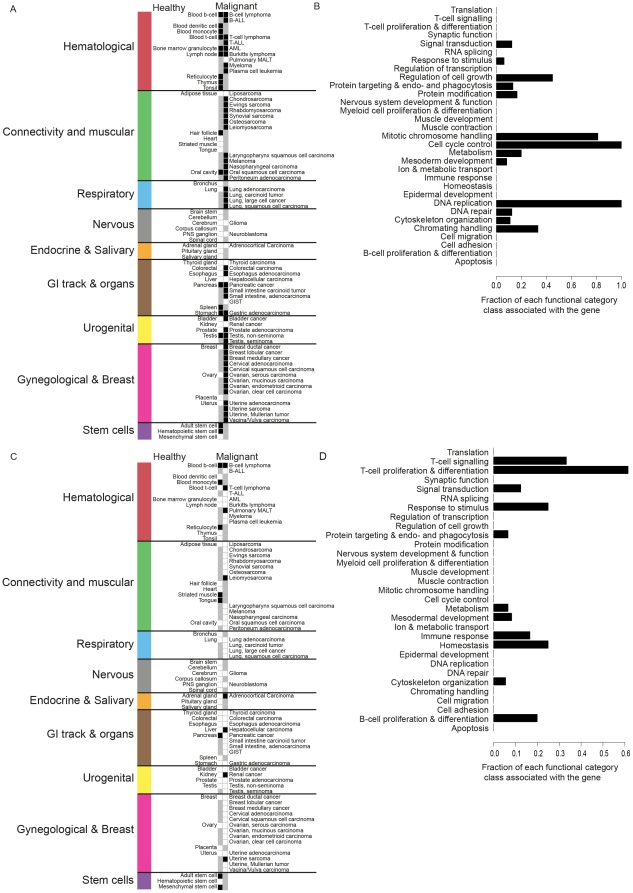
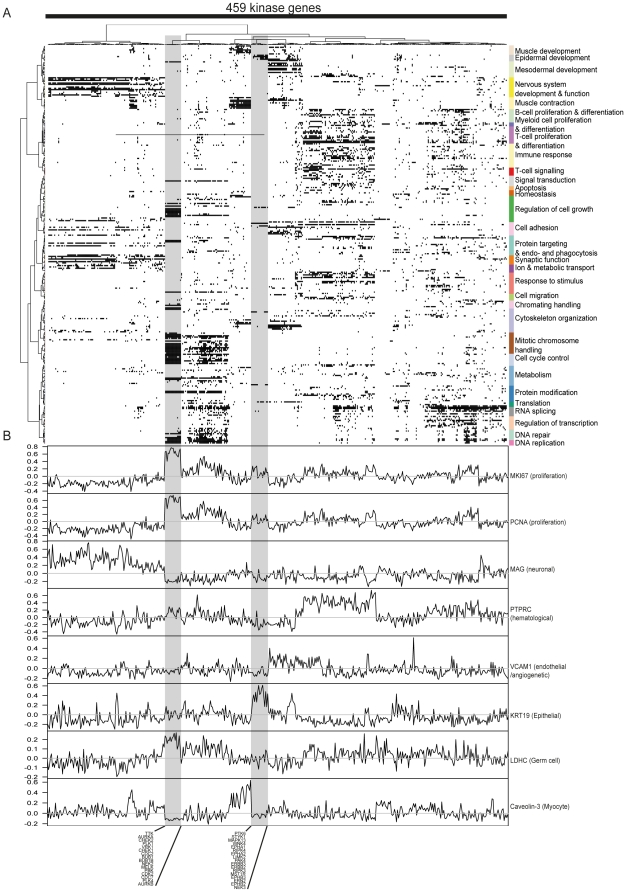
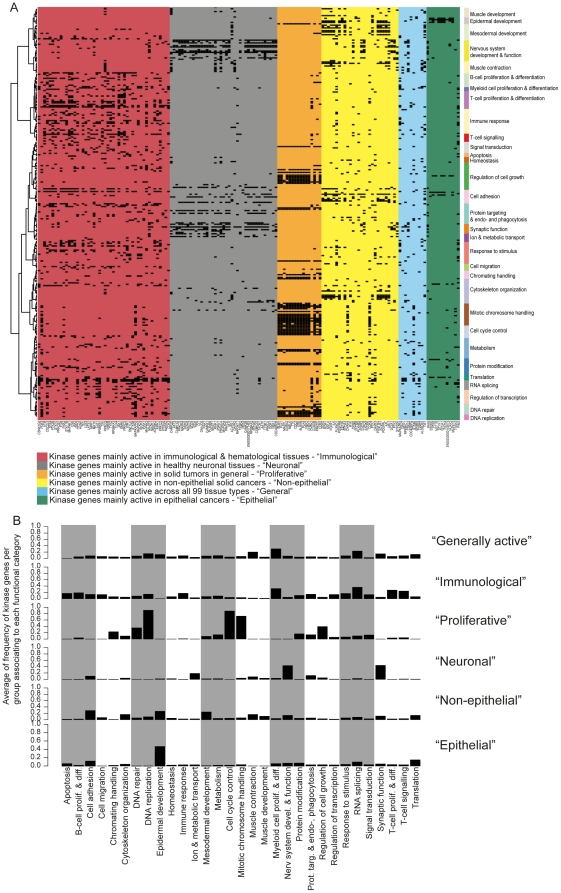
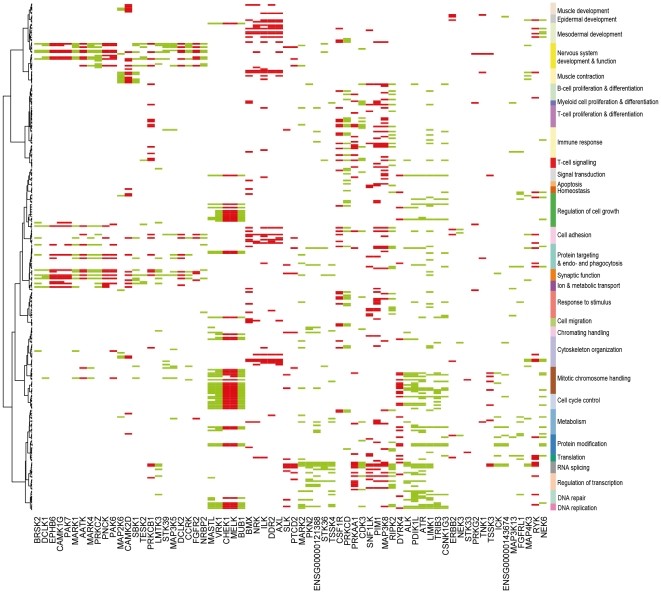
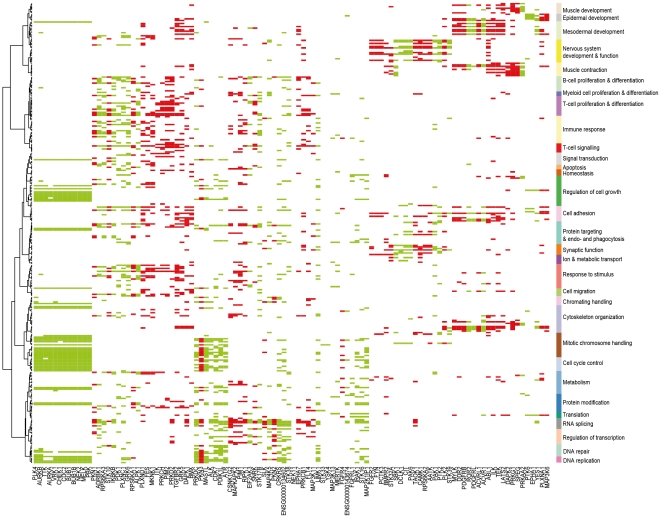
Kinases play key roles in cell signaling and represent major targets for drug development, but the regulation of their activation and their associations with health and disease have not been systematically analyzed. Here, we carried out a bioinformatic analysis of the expression levels of 459 human kinase genes in 5681 samples consisting of 44 healthy and 55 malignant human tissues. Defining the tissues where the kinase genes were transcriptionally active led to a functional genomic taxonomy of the kinome and a classification of human tissues and disease types based on the similarity of their kinome gene expression. The co-expression network around each of the kinase genes was defined in order to determine the functional context, i.e. the biological processes that were active in the cells and tissues where the kinase gene was expressed. Strong associations for individual kinases were found for mitosis (69 genes, including AURKA and BUB1), cell cycle control (73 genes, including PLK1 and AURKB), DNA repair (49 genes, including CHEK1 and ATR), immune response (72 genes, including MATK), neuronal (131 genes, including PRKCE) and muscular (72 genes, including MYLK2) functions. We then analyzed which kinase genes gain or lose transcriptional activity in the development of prostate and lung cancers and elucidated the functional associations of individual cancer associated kinase genes. In summary, we report here a systematic classification of kinases based on the bioinformatic analysis of their expression in human tissues and diseases, as well as grouping of tissues and tumor types according to the similarity of their kinome transcription.
激酶在细胞信号转导中发挥着关键作用,是药物开发的主要靶点,但它们的激活调控及其与健康和疾病的关系尚未得到系统分析。在这里,我们对 459 个人类激酶基因在 5681 个样本中的表达水平进行了生物信息学分析,这些样本包括 44 个健康组织和 55 个恶性组织。定义激酶基因转录活跃的组织导致了对激酶组的功能基因组分类,并基于激酶组基因表达的相似性对人类组织和疾病类型进行了分类。为了确定功能背景,即激酶基因表达的细胞和组织中活跃的生物学过程,定义了每个激酶基因周围的共表达网络。发现个别激酶与有丝分裂(69 个基因,包括 AURKA 和 BUB1)、细胞周期控制(73 个基因,包括 PLK1 和 AURKB)、DNA 修复(49 个基因,包括 CHEK1 和 ATR)、免疫反应(72 个基因,包括 MATK)、神经元(131 个基因,包括 PRKCE)和肌肉(72 个基因,包括 MYLK2)功能密切相关。然后,我们分析了哪些激酶基因在前列腺癌和肺癌的发生过程中获得或失去转录活性,并阐明了个别癌症相关激酶基因的功能关联。总之,我们在这里报告了一种基于人类组织和疾病中激酶表达的生物信息学分析的激酶系统分类,以及根据激酶组转录的相似性对组织和肿瘤类型进行分组的方法。