College of Bioinformatics Science and Technology, Harbin Medical University, Harbin, China.
PLoS One. 2010 Dec 3;5(12):e14219. doi: 10.1371/journal.pone.0014219.
Eukaryotic transcription is accompanied by combinatorial chromatin modifications that serve as functional epigenetic markers. Composition of chromatin modifications specifies histone codes that regulate the associated gene. Discovering novel chromatin regulatory relationships are of general interest.
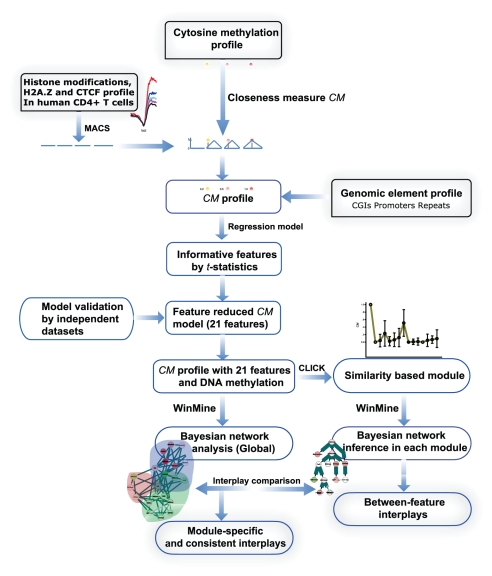
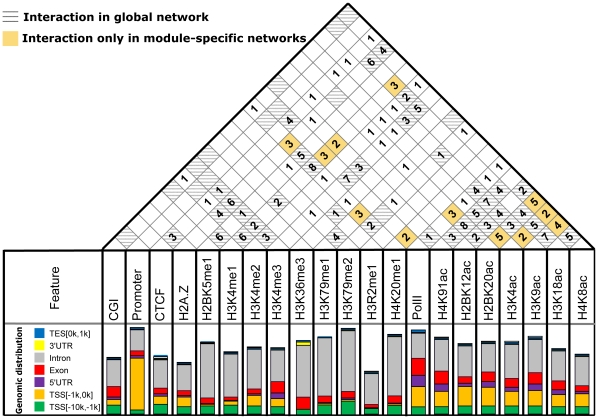
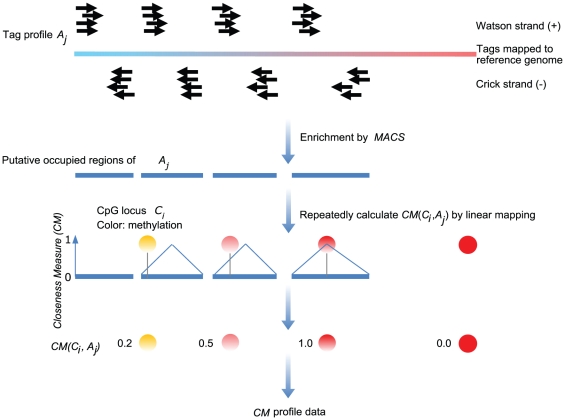
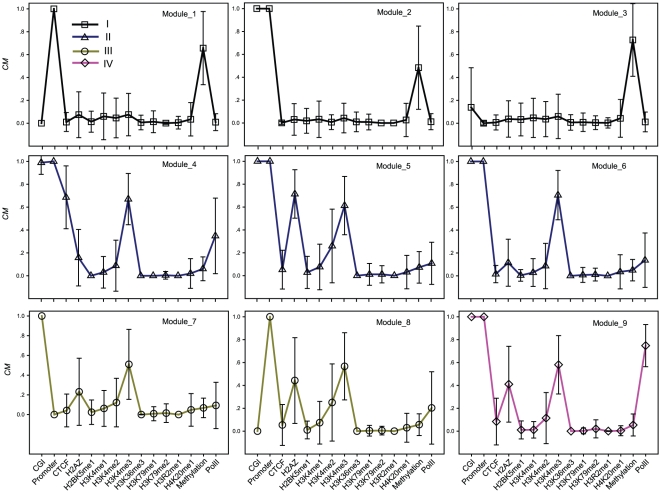
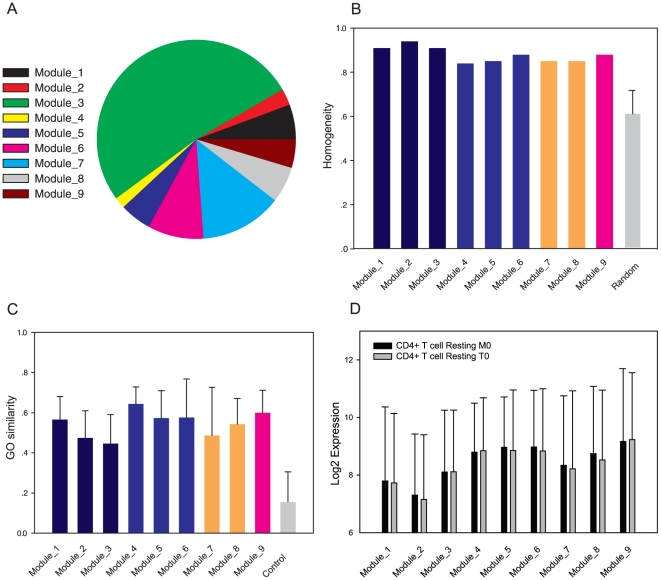
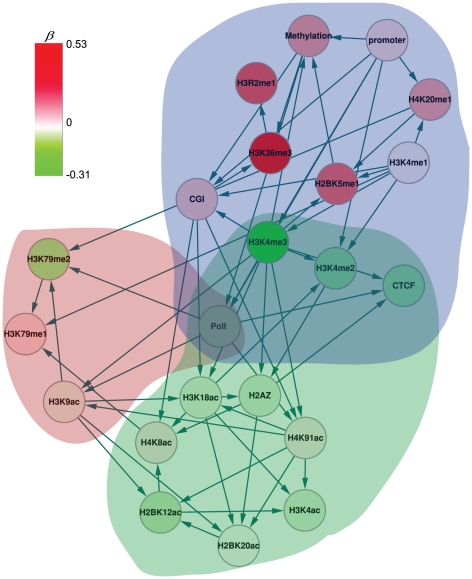
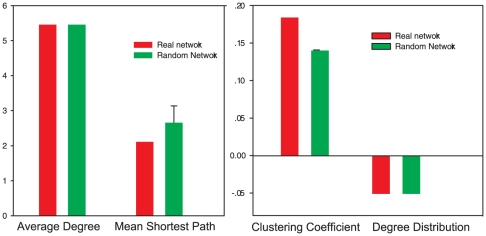
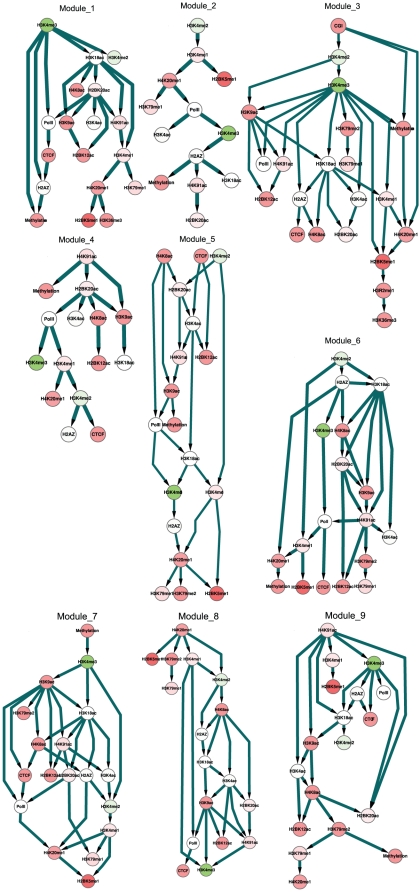
METHODOLOGY/PRINCIPAL FINDINGS: Based on the premise that the interaction of chromatin modifications is hypothesized to influence CpG methylation, we present a closeness measure to characterize the regulatory interactions of epigenomic features. The closeness measure is applied to genome-wide CpG methylation and histone modification datasets in human CD4+T cells to select a subset of potential features. To uncover epigenomic and genomic patterns, CpG loci are clustered into nine modules associated with distinct chromatin and genomic signatures based on terms of biological function. We then performed Bayesian network inference to uncover inherent regulatory relationships from the feature selected closeness measure profile and all nine module-specific profiles respectively. The global and module-specific network exhibits topological proximity and modularity. We found that the regulatory patterns of chromatin modifications differ significantly across modules and that distinct patterns are related to specific transcriptional levels and biological function. DNA methylation and genomic features are found to have little regulatory function. The regulatory relationships were partly validated by literature reviews. We also used partial correlation analysis in other cells to verify novel regulatory relationships.
CONCLUSIONS/SIGNIFICANCE: The interactions among chromatin modifications and genomic elements characterized by a closeness measure help elucidate cooperative patterns of chromatin modification in transcriptional regulation and help decipher complex histone codes.
真核转录伴随着组合性染色质修饰,这些修饰作为功能性表观遗传标记。染色质修饰的组成指定了调节相关基因的组蛋白密码。发现新的染色质调控关系具有普遍意义。
方法/主要发现:基于染色质修饰的相互作用被假设会影响 CpG 甲基化的前提,我们提出了一种接近度度量来描述表观遗传特征的调控相互作用。接近度度量应用于人类 CD4+T 细胞的全基因组 CpG 甲基化和组蛋白修饰数据集,以选择一组潜在的特征。为了揭示表观基因组和基因组模式,CpG 位点根据生物学功能的术语聚类为九个与不同染色质和基因组特征相关的模块。然后,我们分别使用贝叶斯网络推断从特征选择的接近度度量谱和所有九个模块特有的谱中揭示内在的调控关系。全局和模块特有的网络表现出拓扑接近性和模块性。我们发现,染色质修饰的调控模式在不同的模块中存在显著差异,并且不同的模式与特定的转录水平和生物学功能相关。DNA 甲基化和基因组特征的调控作用很小。调控关系部分通过文献综述得到验证。我们还使用其他细胞中的偏相关分析来验证新的调控关系。
结论/意义:由接近度度量来表征的染色质修饰和基因组元件之间的相互作用有助于阐明转录调控中染色质修饰的协同模式,并有助于破译复杂的组蛋白密码。