Department of Chemical Engineering, University of Michigan, Ann Arbor, Michigan, United States of America.
PLoS One. 2010 Dec 13;5(12):e14029. doi: 10.1371/journal.pone.0014029.
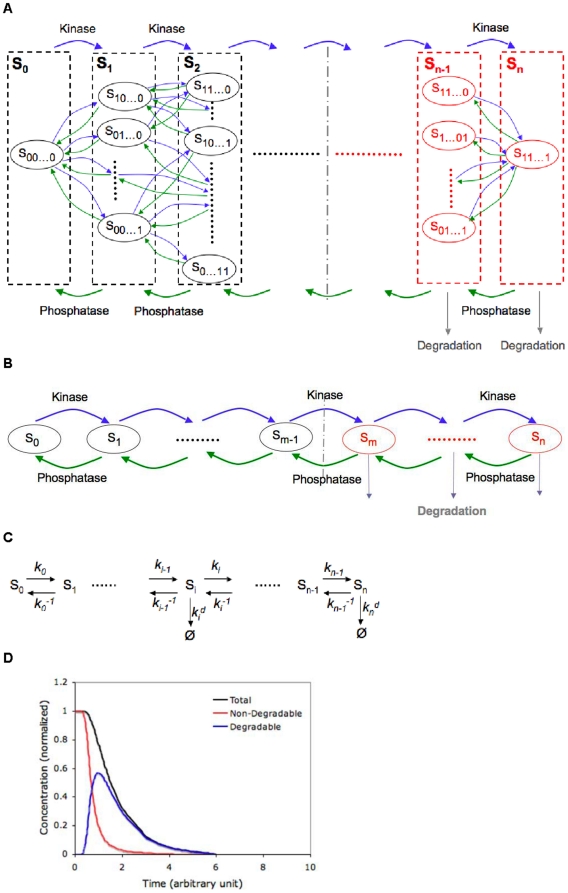
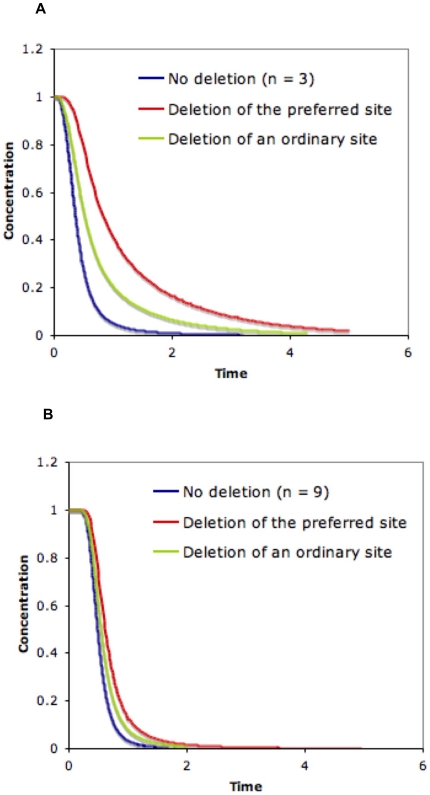
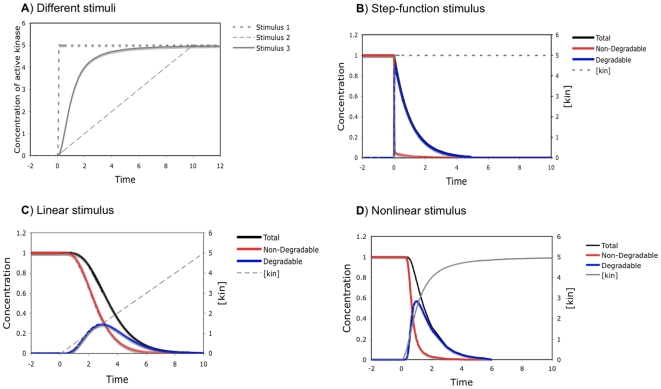
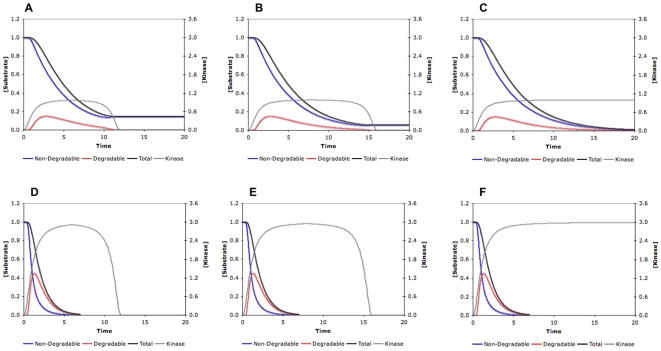
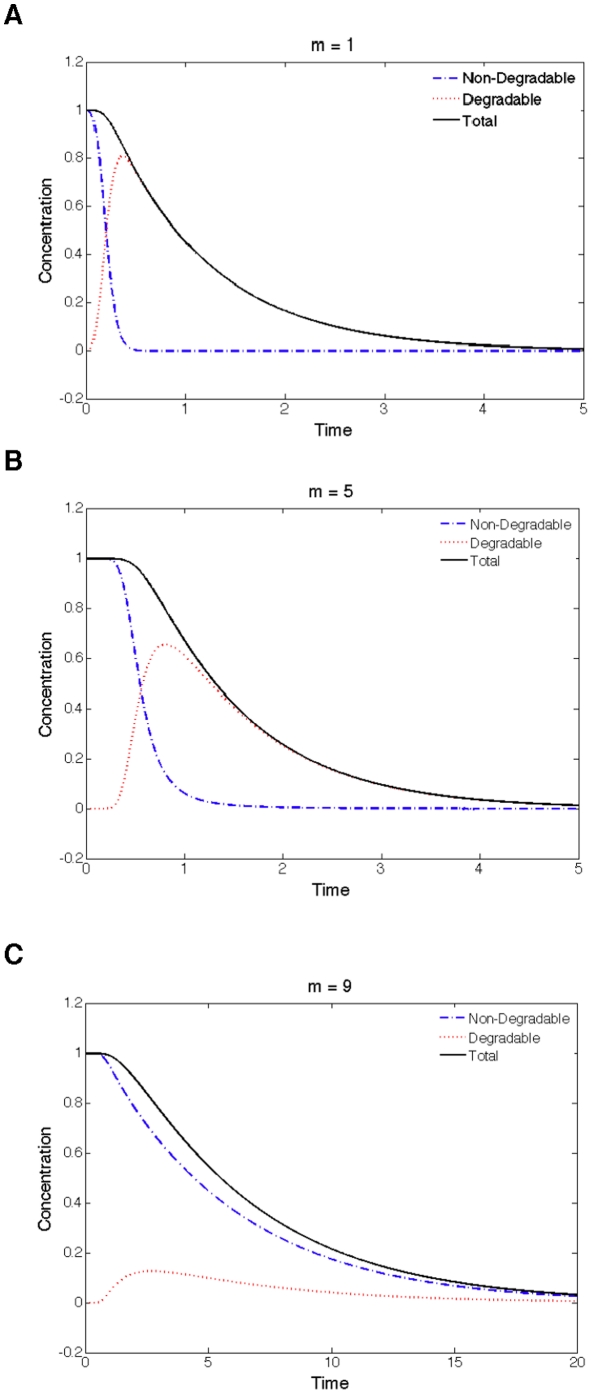
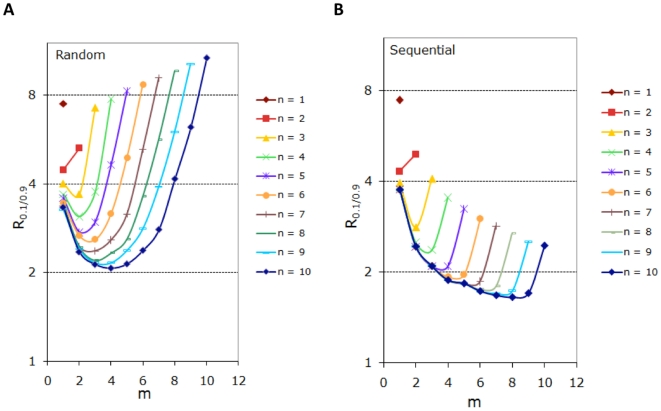
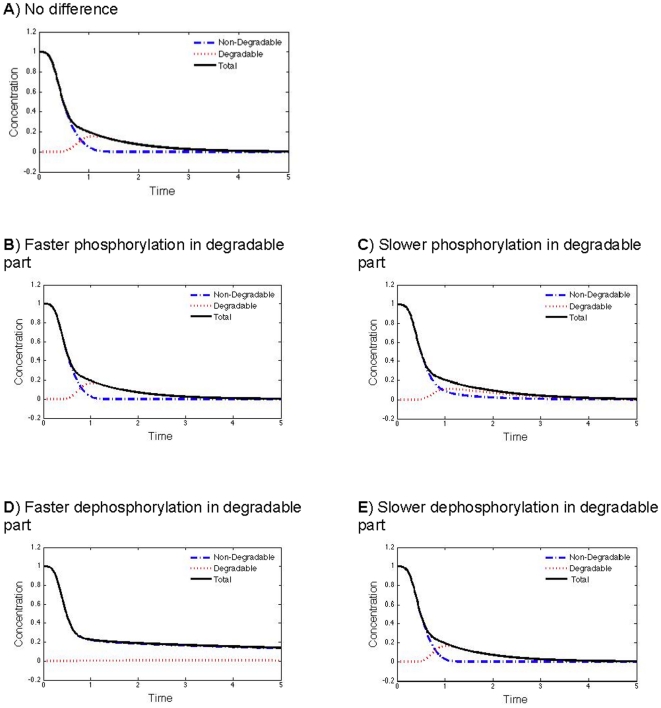
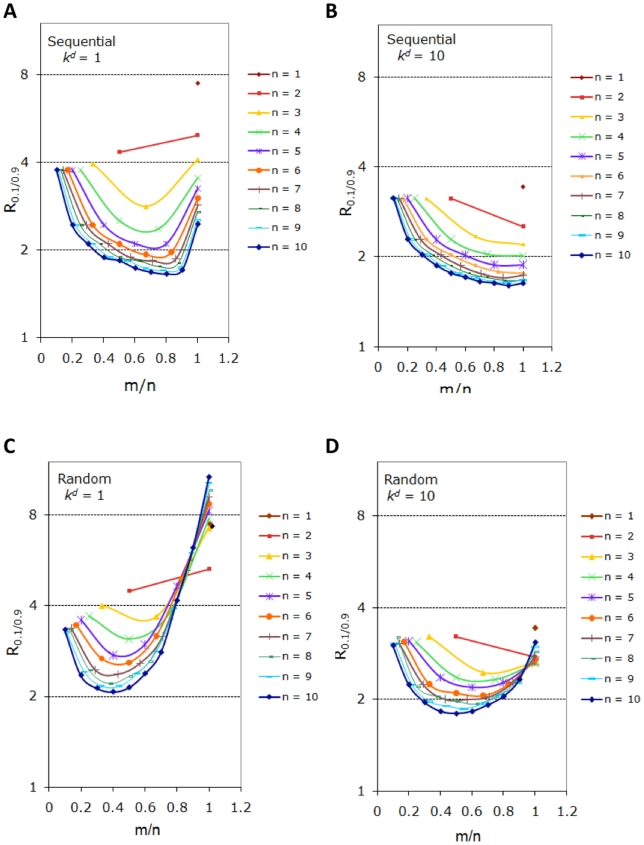
Phosphorylation-triggered degradation is a common strategy for elimination of regulatory proteins in many important cell signaling processes. Interesting examples include cyclin-dependent kinase inhibitors such as p27 in human and Sic1 in yeast, which play crucial roles during the G1/S transition in the cell cycle. In this work, we have modeled and analyzed the dynamics of multisite-phosphorylation-triggered protein degradation systematically. Inspired by experimental observations on the Sic1 protein and a previous intriguing theoretical conjecture, we develop a model to examine in detail the degradation dynamics of a protein featuring multiple phosphorylation sites and a threshold site number for elimination in response to a kinase signal. Our model explains the role of multiple phosphorylation sites, compared to a single site, in the regulation of protein degradation. A single-site protein cannot convert a graded input of kinase increase to much sharper output, whereas multisite phosphorylation is capable of generating a highly switch-like temporal profile of the substrate protein with two characteristics: a temporal threshold and rapid decrease beyond the threshold. We introduce a measure termed temporal response coefficient to quantify the extent to which a response in the time domain is switch-like and further investigate how this property is determined by various factors including the kinase input, the total number of sites, the threshold site number for elimination, the order of phosphorylation, the kinetic parameters, and site preference. Some interesting and experimentally verifiable predictions include that the non-degradable fraction of the substrate protein exhibits a more switch-like temporal profile; a sequential system is more switch-like, while a random system has the advantage of increased robustness; all the parameters, including the total number of sites, the threshold site number for elimination and the kinetic parameters synergistically determine the exact extent to which the degradation profile is switch-like. Our results suggest design principles for protein degradation switches which might be a widespread mechanism for precise regulation of cellular processes such as cell cycle progression.
磷酸化触发的降解是许多重要细胞信号过程中消除调节蛋白的常见策略。有趣的例子包括细胞周期依赖性激酶抑制剂,如人类中的 p27 和酵母中的 Sic1,它们在细胞周期的 G1/S 转换中发挥关键作用。在这项工作中,我们系统地对多位点磷酸化触发的蛋白质降解动力学进行了建模和分析。受 Sic1 蛋白的实验观察和之前一个有趣的理论推测的启发,我们开发了一个模型来详细研究具有多个磷酸化位点和阈值位点数量的蛋白质的降解动力学,以响应激酶信号。我们的模型解释了与单个位点相比,多个磷酸化位点在蛋白质降解调节中的作用。与单个位点相比,单个位点的蛋白质不能将激酶增加的梯度输入转换为更尖锐的输出,而多位点磷酸化能够产生具有两个特征的底物蛋白的高度开关样时间分布:时间阈值和超过阈值后的快速下降。我们引入了一个称为时间响应系数的度量来量化时间域响应的开关样程度,并进一步研究了这个性质如何由各种因素决定,包括激酶输入、位点总数、消除的阈值位点数量、磷酸化顺序、动力学参数和位点偏好。一些有趣的和可以通过实验验证的预测包括:底物蛋白的不可降解部分表现出更开关样的时间分布;顺序系统更具开关样性,而随机系统具有增加鲁棒性的优势;所有参数,包括位点总数、消除的阈值位点数量和动力学参数,协同决定降解谱的开关样程度。我们的结果为蛋白质降解开关提供了设计原则,这可能是细胞过程(如细胞周期进程)精确调控的广泛机制。