Molecular and Human Genetics, Baylor College of Medicine, Houston, Texas, United States of America.
PLoS One. 2010 Dec 20;5(12):e15687. doi: 10.1371/journal.pone.0015687.
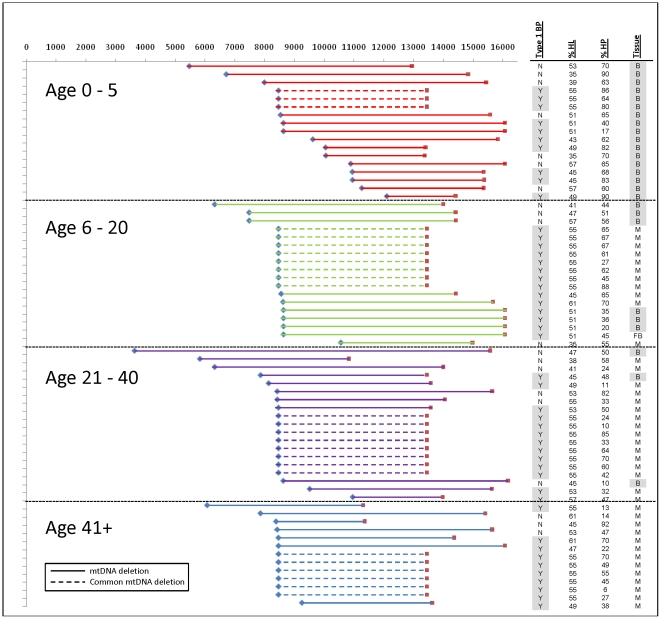
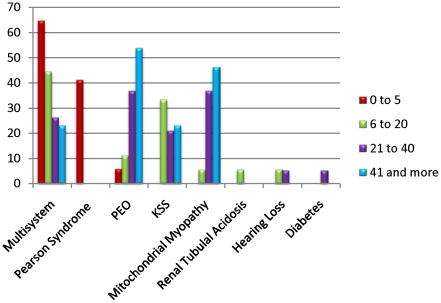
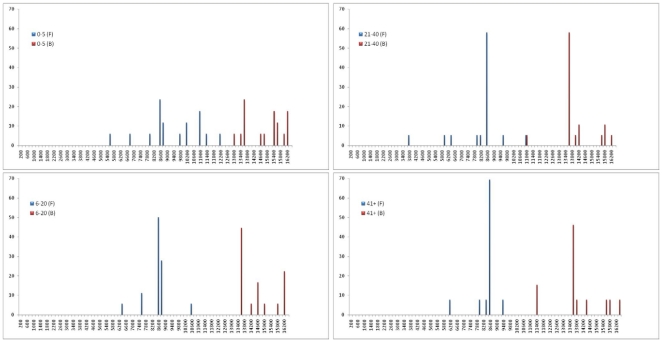
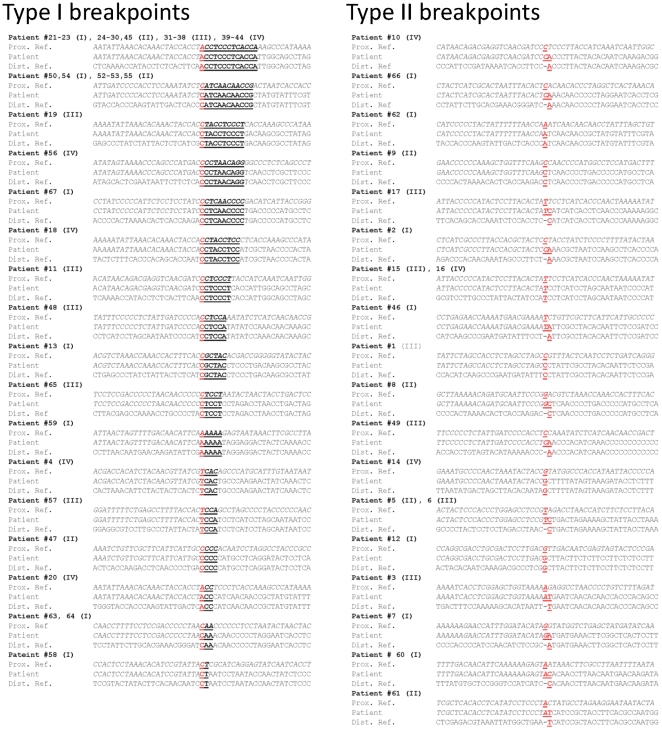
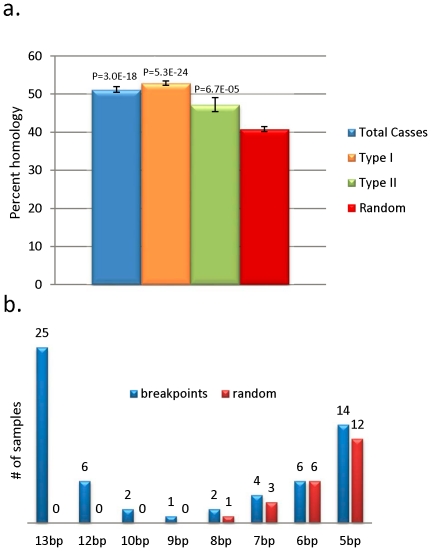
Mitochondrial DNA (mtDNA) deletions are a common cause of mitochondrial disorders. Large mtDNA deletions can lead to a broad spectrum of clinical features with different age of onset, ranging from mild mitochondrial myopathies (MM), progressive external ophthalmoplegia (PEO), and Kearns-Sayre syndrome (KSS), to severe Pearson syndrome. The aim of this study is to investigate the molecular signatures surrounding the deletion breakpoints and their association with the clinical phenotype and age at onset. MtDNA deletions in 67 patients were characterized using array comparative genomic hybridization (aCGH) followed by PCR-sequencing of the deletion junctions. Sequence homology including both perfect and imperfect short repeats flanking the deletion regions were analyzed and correlated with clinical features and patients' age group. In all age groups, there was a significant increase in sequence homology flanking the deletion compared to mtDNA background. The youngest patient group (<6 years old) showed a diffused pattern of deletion distribution in size and locations, with a significantly lower sequence homology flanking the deletion, and the highest percentage of deletion mutant heteroplasmy. The older age groups showed rather discrete pattern of deletions with 44% of all patients over 6 years old carrying the most common 5 kb mtDNA deletion, which was found mostly in muscle specimens (22/41). Only 15% (3/20) of the young patients (<6 years old) carry the 5 kb common deletion, which is usually present in blood rather than muscle. This group of patients predominantly (16 out of 17) exhibit multisystem disorder and/or Pearson syndrome, while older patients had predominantly neuromuscular manifestations including KSS, PEO, and MM. In conclusion, sequence homology at the deletion flanking regions is a consistent feature of mtDNA deletions. Decreased levels of sequence homology and increased levels of deletion mutant heteroplasmy appear to correlate with earlier onset and more severe disease with multisystem involvement.
线粒体 DNA(mtDNA)缺失是线粒体疾病的常见原因。大片段 mtDNA 缺失可导致不同发病年龄的广泛临床特征,从轻度线粒体肌病(MM)、进行性眼外肌麻痹(PEO)和 Kearns-Sayre 综合征(KSS),到严重的 Pearson 综合征。本研究旨在探讨缺失断点周围的分子特征及其与临床表型和发病年龄的关系。使用阵列比较基因组杂交(aCGH)对 67 例患者的 mtDNA 缺失进行了特征分析,然后对缺失连接点进行 PCR 测序。分析了包括缺失区域侧翼的完全和不完全短重复在内的序列同源性,并与临床特征和患者年龄组相关联。在所有年龄组中,与 mtDNA 背景相比,缺失侧翼的序列同源性显著增加。年龄最小的患者组(<6 岁)显示出大小和位置分布弥散的缺失模式,缺失侧翼的序列同源性显著降低,且缺失突变异质体的百分比最高。年龄较大的年龄组显示出离散的缺失模式,6 岁以上的所有患者中有 44%携带最常见的 5kb mtDNA 缺失,这种缺失主要存在于肌肉标本中(22/41)。年龄较小的患者(<6 岁)中只有 15%(3/20)携带常见的 5kb 缺失,这种缺失通常存在于血液中而不是肌肉中。这组患者主要(17 例中有 16 例)表现为多系统疾病和/或 Pearson 综合征,而年龄较大的患者主要表现为神经肌肉表现,包括 KSS、PEO 和 MM。总之,缺失侧翼区域的序列同源性是 mtDNA 缺失的一个一致特征。序列同源性降低和缺失突变异质体增加似乎与发病年龄更早、多系统受累且病情更严重相关。