Department of Life Sciences, National Cheng Kung University, Tainan, Taiwan.
BMC Plant Biol. 2011 Jan 6;11:3. doi: 10.1186/1471-2229-11-3.
Phalaenopsis orchids are popular floral crops, and development of new cultivars is economically important to floricultural industries worldwide. Analysis of orchid genes could facilitate orchid improvement. Bacterial artificial chromosome (BAC) end sequences (BESs) can provide the first glimpses into the sequence composition of a novel genome and can yield molecular markers for use in genetic mapping and breeding.
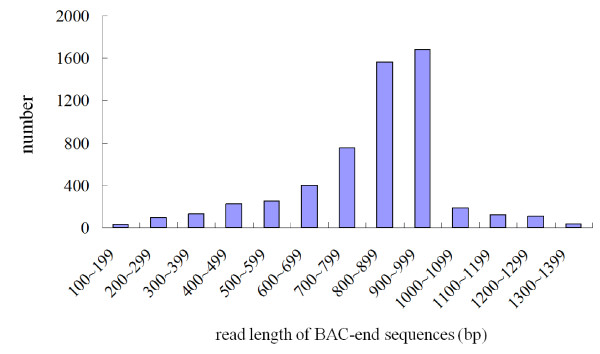
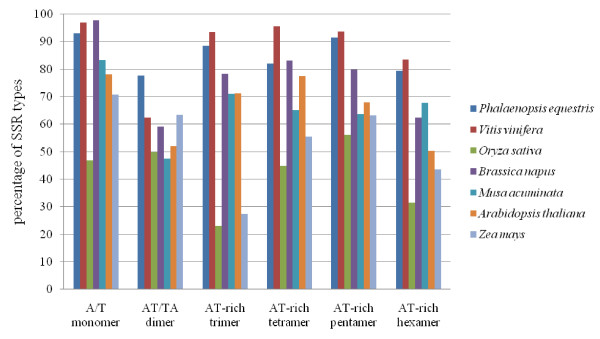
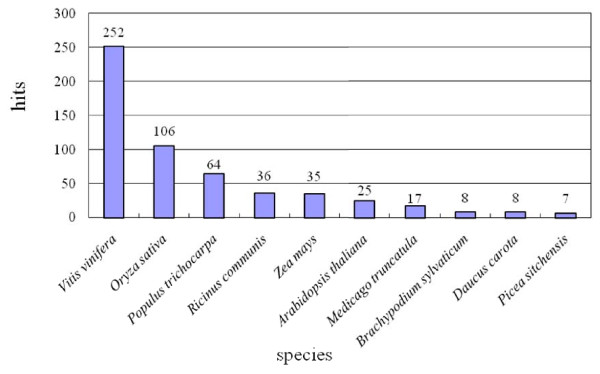
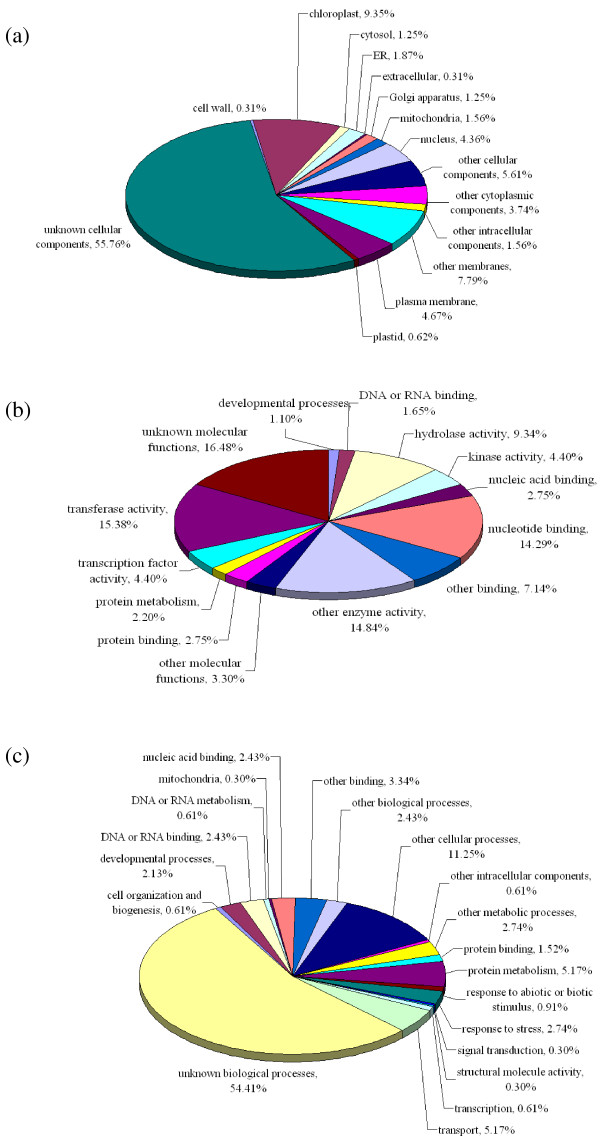
We used two BAC libraries (constructed using the BamHI and HindIII restriction enzymes) of Phalaenopsis equestris to generate pair-end sequences from 2,920 BAC clones (71.4% and 28.6% from the BamHI and HindIII libraries, respectively), at a success rate of 95.7%. A total of 5,535 BESs were generated, representing 4.5 Mb, or about 0.3% of the Phalaenopsis genome. The trimmed sequences ranged from 123 to 1,397 base pairs (bp) in size, with an average edited read length of 821 bp. When these BESs were subjected to sequence homology searches, it was found that 641 (11.6%) were predicted to represent protein-encoding regions, whereas 1,272 (23.0%) contained repetitive DNA. Most of the repetitive DNA sequences were gypsy- and copia-like retrotransposons (41.9% and 12.8%, respectively), whereas only 10.8% were DNA transposons. Further, 950 potential simple sequence repeats (SSRs) were discovered. Dinucleotides were the most abundant repeat motifs; AT/TA dimer repeats were the most frequent SSRs, representing 253 (26.6%) of all identified SSRs. Microsynteny analysis revealed that more BESs mapped to the whole-genome sequences of poplar than to those of grape or Arabidopsis, and even fewer mapped to the rice genome. This work will facilitate analysis of the Phalaenopsis genome, and will help clarify similarities and differences in genome composition between orchids and other plant species.
Using BES analysis, we obtained an overview of the Phalaenopsis genome in terms of gene abundance, the presence of repetitive DNA and SSR markers, and the extent of microsynteny with other plant species. This work provides a basis for future physical mapping of the Phalaenopsis genome and advances our knowledge thereof.
蝴蝶兰是一种受欢迎的花卉作物,培育新品种对全球花卉产业具有重要的经济意义。对兰花基因的分析可以促进兰花的改良。细菌人工染色体(BAC)末端序列(BES)可以提供对新基因组序列组成的初步了解,并可产生用于遗传图谱构建和育种的分子标记。
我们使用 Phalaenopsis equestris 的两个 BAC 文库(分别使用 BamHI 和 HindIII 限制酶构建),从 2920 个 BAC 克隆中生成了末端序列(BamHI 和 HindIII 文库的成功率分别为 71.4%和 28.6%),共产生了 5535 个 BES,代表 4.5Mb,约占 Phalaenopsis 基因组的 0.3%。经修剪的序列大小从 123 到 1397 个碱基对(bp)不等,平均编辑读取长度为 821bp。当这些 BES 进行序列同源性搜索时,发现其中 641 个(11.6%)预测为编码蛋白的区域,而 1272 个(23.0%)含有重复 DNA。大多数重复 DNA 序列是 gypsy 和 copia 样反转录转座子(分别为 41.9%和 12.8%),而只有 10.8%是 DNA 转座子。此外,还发现了 950 个潜在的简单序列重复(SSR)。二核苷酸是最丰富的重复基序;AT/TA 二聚体重复是最常见的 SSR,占所有鉴定 SSR 的 253 个(26.6%)。微同线性分析显示,更多的 BES 映射到杨树的全基因组序列,而不是葡萄或拟南芥的全基因组序列,映射到水稻基因组的则更少。这项工作将有助于分析蝴蝶兰基因组,并帮助阐明兰花和其他植物物种在基因组组成上的相似性和差异。
通过 BES 分析,我们从基因丰度、重复 DNA 和 SSR 标记的存在以及与其他植物物种的微同线性程度等方面对蝴蝶兰基因组进行了概述。这项工作为蝴蝶兰基因组的物理图谱构建提供了基础,并增进了我们对其的了解。